Startseite » 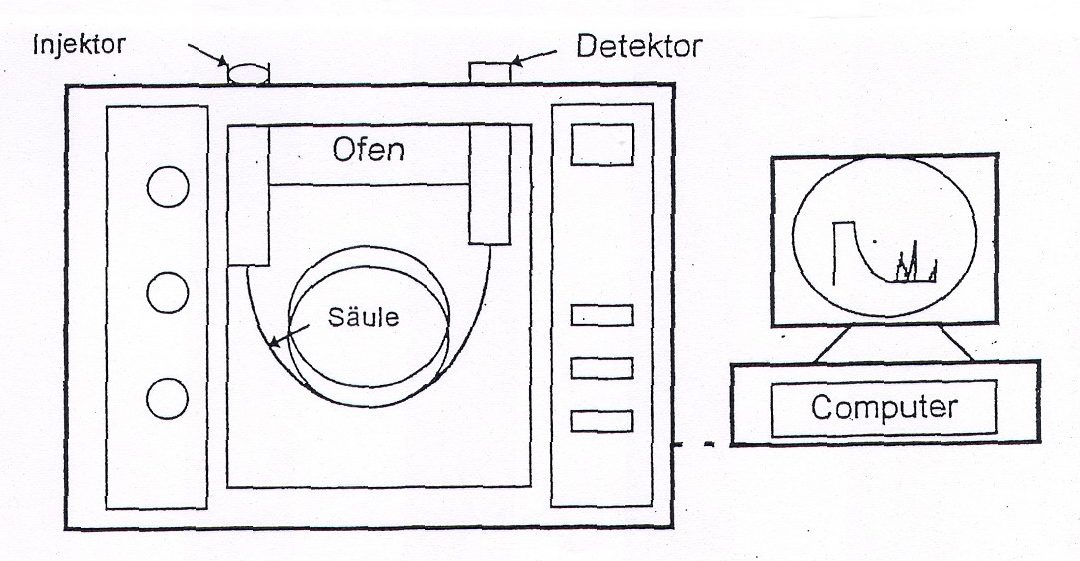
Allgemeiner Leitfaden für Neueinsteiger
Auf den folgenden Seiten sind einige theoretische Grundlagen und Handlungsanweisungen für das erste Arbeiten mit einem GC beschrieben.
Das soll nicht das Lesen von Lehrbüchern ersetzen, vor allem kann es nicht die Handhabung der Geräte einzelner Hersteller erläutern. Im Abschnitt 15 sind Internetquellen und eine Liste mit GC-Büchern zu finden.
Was ist überhaupt GC?
GC
1950 wurde die GC von Erika Cremer als Gas-Adsorptionschromatographie vorgestellt. Die Entwicklung ging dann Schlag auf Schlag. Schon 1952 wurde die Gas-Verteilungschromatographie entdeckt und beschrieben und 1961 gab es schon 40 kommerzielle GC-Hersteller.
In den 1980‘ern hat die GC durch die rasant fortschreitende Computertechnologie noch mal einen riesigen Schub erlebt. Heute wird vor allem technisch an der dazu gehörenden Instrumenten-steuerung, an Kopplungstechniken und Datenverarbeitung optimiert.
Voraussetzung
Die Gaschromatographie beruht, wie andere chromatographische Verfahren, auf Verteilung und/oder Adsorption. Voraussetzung ist, dass die zu untersuchenden Substanzen gasförmig vorliegen oder sich durch Verdampfen bei den überwiegenden Anwendungen möglichst unzersetzt in den gasförmigen Zustand überführen lassen.
Mobile Phase Trennsäule Ofen
Die gasförmigen oder verdampften Substanzen werden mit dem Trägergasstrom (=mobile Phase) durch eine Trennsäule transportiert. Diese befindet sich in einem Ofen und kann auf Temperaturen bis zu ca. 450°C beheizt werden.
Trägergase
Typische Trägergase für die Gaschromatographie sind Helium, Stickstoff und Wasserstoff. Die Wahl der Trägergase richtet sich nach dem Detektor, der Trennleistung und den Kosten.
Stationäre Phase
Die stationäre Phase kann entweder eine sogenannte gepackte Säule sein (Glasrohr gefüllt mit Säulenmaterial) oder eine Kapillarsäule. Diese Kapillarsäulen (Kapillare = fused silica (Quarz), Mantel = Polyimid, braun) können unterschiedliche Trennmaterialien enthalten. Moderne GC-Kapillarsäulen sind innen häufig mit einem hoch vernetzten Film unterschiedlicher Dicke beschichtet.
GLC/GSC
Je nach Aggregatzustand der stationären Phase unterscheidet man die Gas-Flüssig-Chromatographie (GLC) und die Gas-Fest-Chromatographie (GSC).
Probenaufgabe
Bei der Gaschromatographie verwendet man, abhängig von der Art der Probe, der Dimension der Säule und Probenmenge, unterschiedliche Probeaufgabetechniken.
Um eine Probe auf die Trennsäule zu bringen, muss die Substanz, falls sie nicht schon gasförmig vorliegt, im Injektor verdampft werden.
Injektor
Prinzipiell wird hier zwischen Injektoren für gepackte Säulen und Injektoren für Kapillarsäulen unterschieden.
Splitting
Bei den Injektoren für Kapillarsäulen wird, wegen der geringen Belastbarkeit dieses Säulentyps, meist eine Probenaufgabe unter Stromteilung (splitting) eingesetzt, die so genannte Splitaufgabetechnik.
Splitverhältnis
Das Splitverhältnis ist definiert als Quotient des Trägergasstromes durch die Säule und dem Trägergasfluss durch das Splitventil.
Auswertung
Die einzelnen Probekomponenten werden dann am Ende der Trennsäule von einem Detektor registriert, dieser gibt die Informationen an die Auswerteeinheit weiter, das Ergebnis ist ein Chromatogramm.
Die Anzahl der Peaks entspricht der Anzahl der aufgetrennten (!) Probekomponenten, die Fläche ist deren Menge oder Masse proportional.
Für eine quantitative Auswertung werden Standards eingesetzt. Im optimalen Fall arbeitet man bei der GC mit externen Standards und erstellt eine Kalibrierkurve. Häufigere Auswertetechnik ist jedoch die Auswertung über interne Standards (z. B. wenn die zu bestimmende Substanz selbst nicht zur Verfügung steht oder die Standardaddition z. B. wenn ein größerer Matrixeinfluss vorliegt und man mit einem Wiederfindungsproblem oder mit Verlusten während der Injektion rechnen muss).
Detektor
Für die Detektion der getrennten Substanzen können verschiedene Detektoren eingesetzt werden.
WLD
Der älteste Detektor ist der Wärmeleitfähigkeitsdetektor (WLD), der den Unterschied der Wärmeleitfähigkeit von Trägergas zu Trägergas mit Probe durch Widerstandsmessung (Wheatstone´sche Brücke) misst.
FID
Heute wird in der Routine am häufigsten der Flammenionisationsdetektor (FID) eingesetzt. Der FID verbrennt und ionisiert organische Kohlenwasserstoffe in einer Flamme. Die Erhöhung des Stromflusses beim Ankommen der ionisierten organischen Verbindungen wird registriert.
Die Auswertung der Detektorsignale und die Einstellung/Steuerung der Gasflüsse, Ventile und Temperaturen im Injektor, Säulenofen und Detektor wird heute meist über einen PC mit einer Spezialsoftware vorgenommen, die über eine Steuerungssoftware die Methoden an den GC weitergeben.
MS
Seit den 1990er Jahren wird immer mehr ein Massenspektrometer als massenselektiver Detektor verwendet (GC/MS-Kopplung). Durch die selektive Fragmentierung der zu trennenden Komponenten können spezifische Informationen über die Struktur der Moleküle erhalten werden.
Aus welchen Teilen besteht ein GC?
Aufbau
- einer oder mehrerer Gasflaschen mit Reduzierventilen für Trägergase und Brenngase
- einem Probenaufgabesystem (evtl. inkl. Autosampler)
- einer Säule, die sich in einem temperierbaren Ofen befindet
- einem Detektor zur Registrierung der Probenkomponenten
- und zumeist einem Rechner zum Steuern der Ventile, Temperatur und zur Aufnahme der erzeugten Signale
Gasversorgung
Gase
Für den Transport der Probenkomponenten durch die Säule zum Detektor wird ein Trägergas benötigt. Dieses Trägergas wird entweder durch eine Druckgasflasche (mit 100 – 200 bar) oder einen Gasgenerator dem GC zugeführt. Vor dem GC müssen noch Reduzierventile und Druckregler angebracht sein, um den Gasdruck auf ca. 2 – 3 atm (5 bar) herunter zu regeln. Typische Trägergase sind Helium, Argon oder auch Wasserstoff.
Reinheit
Die Gase sollten über größtmögliche Reinheit verfügen.
Bei der Analytik im Spurenbereich sollte man auf alle Fälle die Reinheit 5,5 wählen (=99,9995% rein).
Gasreinigung
Bei schlechterer Reinheit sollte man so genannte Gasreinigungssysteme (z. B. Kartuschen mit Aktivkohle) in die Gasversorgung vor dem GC einbauen.
Der eingestellte Gasfluss sollte bei gepackten Säulen zwischen 15- 60 ml/min liegen.
Septumpurge
Der Septumpurge (ein minimaler Gasfluss unterhalb des Septums und oberhalb des Liners) sorgt dafür, dass keine Kontaminationen aus dem Septum auf die Säule gelangen.
Liner
Ein Liner oder auch „Glasliner“ stellt die Verbindung zwischen dem Injektionssystem und der Säule her und ist aus inertem Glas.
Probenaufgabe/Injektoren
Injektor
Probenaufgabe
Prinzipiell wird hier zwischen Injektoren für gepackte Säulen und Injektoren für Kapillarsäulen unterschieden.
Bei Injektoren für Kapillarsäulen wird wegen der geringen Belastbarkeit dieses Säulentyps meist eine Probenaufgabe unter Stromteilung (splitting) eingesetzt, die so genannte Splitaufgabetechnik.
Split
Das Splitverhältnis ist definiert als Quotient des Trägergasstromes durch das Splitventil und dem Trägergasfluss durch die Säule, z. B. Splitverhältnis 1:100 bedeutet, dass 1 ml/min durch die Säule strömt und 100 ml/min aus dem Splitventil herausgeleitet wird.
Die Bestimmung dieses Gasflusses kann z. B. manuell mit Blasenzähler oder automatisch über eine elektronische Pneumatik (Gasflussregulierung) erfolgen.
Diese Systeme zur Gasflussregulierung können die Flusskonstanz in der Säule sehr genau regulieren. In der folgenden Tabelle sind die z. Zt. am häufigsten eingesetzten Injektionstechniken aufgeführt:
| Injektortyp: | Säulentyp: | Einspritzmenge: | Proben: |
| Split/Splitlos | Kapillarsäulen | 0,1 – 2 µl | flüssig |
| On-Column | Kapillarsäulen | 0,1 – 10 µl | flüssig |
Die Probenaufgabe erfolgt mit folgenden Systemen:
| Gasproben– oder Dosierstreifen | gasförmig | 0,1 – 10 ml | Gasförmige Proben können gezielt gesammelt und aufgegeben werden. |
| PTV Programmed Temperatur Vaporizer (Kaltaufgabe) | flüssig | 0,1 – > 10 µl | Man erreicht gezielte Verdampfung und hohe Anreicherung, große Injektionsmengen möglich |
Die Detektoren
Für die Detektion der getrennten Substanzen stehen folgende Detektoren zur Verfügung:
| Abk. | Prinzip | engl. Bez. | Anwendung | LOD* | Bereich [g] |
| FID | Flammenionisation | flame ionization | selektiv für alle org. Substanzen | 1 ng | bis ca. 10-9 |
| WLD/ TCD | Wärmeleitfähigkeit | thermal conductivity | universell | 1 µg | bis ca. 10-6 |
| TID / NPD | Thermoionisation | thermo- ionization | selektiv alle P- u. N- haltige Verbindungen (Heteroatome) | 10 pg | bis ca. 10-10 |
| ECD | Elektroneneinfang | electron capture | Halogene u. N–haltige Verbindungen | 1 pg | bis ca. 10-12 |
| MS(D) | Massenspektrometer | mass spectrometer | universell oder spezifisch | bis zu 1 fg | je nach Einstellung |
*Nachweisgrenze („Limit of Detection“)
Die Nachweisgrenze ist neben der Dosiermenge auch von der zu analysierenden Substanz abhängig. Die Nachweisgrenze wird in der GC – wie in anderen chromatographischen Techniken – meist mit dem Signal/Rauschen-Verhältnis 3:1 definiert. Die Bestimmungsgrenze (LOQ = Limit of Quantitation) mit dem Signal/Rauschen-Verhältnis von mindestens 10:1.
In der GC sind die Signale (Peaks) bei üblichen Standardanalysen (z. B. Lösungsmittelbestimmungen) meist sehr schmal und scharf umrissen, so dass hier die Auswertung über automatisierte Auswerteprogramme oft einfacher ist, wie beispielsweise bei der HPLC.
FID
Flammenionisationsdetektor
Der am häufigsten in der GC eingesetzte Detektor ist der FID. Die Einsatzgebiete reichen von Applikationen in der Lebensmittelchemie, Umweltanalytik bis hin zur Analytik von pharmazeutischen Zubereitungen.
Arbeitsweise:
Es wird die elektrische Leitfähigkeit einer Wasserstoffflamme im elektrischen Feld gemessen und wenn eine organische Substanz in die Flamme gelangt, so wird diese verbrannt (=ionisiert) und verändert den Stromfluss proportional zur Probenmenge.
Der FID ist relativ nachweisempfindlich und universell einsetzbar. Da die bei der Signalerzeugung benötigten Ionen hauptsächlich bei der Verbrennung von organischen Kohlenwasserstoffgruppen entstehen, zeigt der FID nicht alle Verbindungen an. Substanzen auf die der FID wenig oder gar nicht anspricht sind: Edelgase, H2, N2, Stickstoffoxide, CO, CCl4 oder andere halogenierte Verbindungen, Siliciumhalogenide, CO2, H2O, CS2, NH3, O2. Im Gegensatz zu einigen anderen Detektoren (z. B. ECD oder WLD) arbeitet der FID destruktiv. Das bedeutet, dass die zu analysierende Probe (hier durch Verbrennung) zerstört wird.
WLD/TCD
Wärmeleitfähigkeitsdetektor:
Dieser universelle und vergleichsweise nachweisunempfindliche Detektor erfasst alle organischen Substanzen. Die Nachweisempfindlichkeit hängt vom eingesetzten Trägergas ab.
Arbeitsweise:
Der von der Säule kommende Gasstrom wird an einem Heizdraht vorbeigeführt. Dieser wird durch das vorbeiströmende Trägergas ständig leicht abgekühlt. Die zu untersuchenden Substanzen haben eine geringere Wärmeleitfähigkeit, als das reine Trägergas, so dass das Messelement in ihrer Gegenwart weniger abgekühlt wird. Die daraus erfolgte Temperaturerhöhung wird als Änderung eines elektrischen Widerstandes gegenüber einem Referenzelement gemessen.
ECD
Elektroneneinfangdetektor (engl.: Electron Capture Detector)
Spezifischer und sehr nachweisempfindlicher Detektor für Halogene und andere Substanzen mit hoher Elektronenaffinität.
Arbeitsweise:
Der Trägergasstrom wird durch eine Ionisationskammer geleitet, in der sich ein ß-Strahler (z. B. 63 NI) befindet. Um einfangbare thermische Elektronen zu erzeugen, wird dieses Trägergas mit ß-Teilchen ionisiert. Der entstehende Elektronenfluss produziert einen kleinen Strom, der dann gemessen wird. Wenn Substanzen mit hoher Elektronenaffinität die Zelle erreichen, fangen sie die freien Elektronen ein, die sonst vom Kollektor gesammelt und einen Strom ergeben würden. Der reduzierte Strom wird dann als Signaländerung (=Peak) registriert.
MS(D)
Massenspektrometer/Massenselektiver Detektor
Je nach Aufnahmemodus universell oder selektiv einsetzbar und nachweisempfindlich.
Arbeitsweise:
Die Substanzen werden in einem Ionisationsraum mit Elektronen beschossen, diese zerfallen auf eine für jede Substanz typische Weise (Fragmente -> Massenspektrum). Die entstandenen Fragmente werden in einem Analysatorraum (z. B. massenselektive Filter und ein Quadrupol) nach dem Verhältnis Masse/Ladung sortiert und durch einen Sekundärelektronenvervielfacher gezählt.
Make-up-Gase
Einige Detektoren benötigen so genannte Make-up-Gase, z. B. FID oder manche WLD’s.
Das ist dann der Fall, wenn der Detektor eigentlich für gepackte Säulen konstruiert wurde, aber jetzt mit Kapillarsäulen eingesetzt wird. Die Nachweisempfindlichkeit wird dann meist nicht bei diesen kleinen Gasflüssen aus der Säule erreicht und man muss dem Detektor noch Gase zuführen. Der Anschluss des Gases ist meist direkt am Detektor. Erkundigen Sie sich, ob Ihr Detektor ein Make-up-Gas benötigt.
Die stationäre Phase /Trennsäulen
Trennsäulen gibt es von vielen Herstellern und gerade als Neueinsteiger muss man sich durch einen Dschungel von unterschiedlichen Bezeichnungen hindurcharbeiten. In den folgenden Tabellen sind einige Grundbegriffe erklärt und auch Beispiele einiger wichtiger Säulen gegeben.
Die Nachweisgrenze ist neben der Dosiermenge auch von der zu analysierenden Substanz abhängig. Die Nachweisgrenze wird in der GC – wie in anderen chromatographischen Techniken – meist mit dem Signal/Rauschen-Verhältnis 3:1 definiert. Die Bestimmungsgrenze (LOQ = Limit of Quantitation) mit dem Signal/Rauschen-Verhältnis von mindestens 10:1.
In der GC sind die Signale (Peaks) bei üblichen Standardanalysen (z. B. Lösungsmittelbestimmungen) meist sehr schmal und scharf umrissen, so dass hier die Auswertung über automatisierte Auswerteprogramme oft einfacher ist, wie beispielsweise bei der HPLC.
Trennsäulen
Es sind zwei prinzipiell unterschiedliche Typen von gaschromatographischen Trennsäulen im Einsatz:
- gepackte Trennsäule
- Kapillartrennsäulen
gepackte Säulen
Gepackte Trennsäulen haben in der Regel eine Länge von ca. 0,5 – 3 m, einen Innendurchmesser von ca. 2 – 4 mm und bestehen aus einem Glas- oder Metallrohr. Sie sind mit einem festen Adsorbens oder Trägermaterial gefüllt (gepackt).
Kapillarsäulen Trennleistung
Kapillartrennsäulen haben Längen von ca. 10 bis über 100 m. Ihr Durchmesser beträgt nur ca. 0,05 bis 0,75 mm. Ihre Trennleistung ist durchschnittlich ca. 10 bis 100 mal größer als die von gepackten Säulen.
Polyimid-Film
Die Kapillaren werden im Allgemeinen aus „synthetischem Quarz“ (engl.: fused silica) hergestellt. Die sehr dünnwandigen Kapillaren sind zur Stabilitätsverbesserung außen mit einem Polyimid-Film überzogen, dadurch werden die Kapillaren sehr flexibel, was einen großen Vorteil in der Handhabung bedeutet.
Strömungsverhältnisse
Die Strömungsverhältnisse bei der Anwendung von Kapillarsäulen sind um ein mindestens 10-faches niedriger als mit gepackten Säulen und die Einspritzmenge ist max. 1/10 so groß.
Glasliner
Im Gegensatz zu den gepackten Säulen ist der Verdampferraum nicht schon in der Säule, sondern in einem inerten Glasliner („Verdampferröhrchen“ aus Spezialglas) vor der Kapillare. Dieser muss für den entsprechenden GC bzw. das Injektionssystem angepasst sein.
Split
Ein großer Teil der injizierten Substanzen, die eine Kapillarsäule überladen könnten, wird über ein Split direkt hinter dem Injektor mit Trägergas aus dem GC herausgeleitet. Damit man vergleichbare Ergebnisse erhält, muss dieser Split während der gesamten Analytik (Standard und Proben) gleich konstant gehalten werden.
Gepackte Säulen sind heute sehr selten im Einsatz, da u.a. durch die viel höheren Bodenzahlen (10 bis 100-fach höher) mit den Kapillarsäulen viel bessere Trennungen erreicht werden können (bessere Effizienz, scharfe Peaks).
Außerdem ist die Auswahl an Trennmaterialien für gepackte Säulen inzwischen recht eingeschränkt.
Die folgende Tabelle gibt einen Überblick über die Unterschiede zwischen gepackten Säulen und Kapillarsäulen:
| Gepackte Säulen | Kapillarsäulen | |
| Material | Glas, Stahl | Quarz (fused –silica mit Polyimidüberzug) |
| Länge | 0,5 – 3 m | 10 – 100 m |
| Innendurchmesser | 2 – 5 mm | 0,05 – 0,75 mm |
| Belastbarkeit | bis ca. 20 µg/Peak | bis ca. 50 ng/Peak |
| Trennleistung/ Bodenzahl | N = max. 5000 | N > 100.000 |
| Stationäre Phase | Inertes Trägermaterial (evtl. mit aufgezogener flüssiger stationärer Phase) | SCOT * = Dünnschichtsäulen WCOT * = Dünnfilmsäulen PLOT * = mikrogepackte Säulen |
*Diese Abkürzung werden in der folgenden Tabelle erklärt
Die folgende Tabelle gibt eine Übersicht über die gebräuchlichen Abkürzungen bei Kapillarsäulen und einige wichtige Eigenschaften.
| Abk.: | engl. Bezeichnung: | Bedeutung: | I.D. [mm] | Filmdicke |
| WCOT | Wall coated open tubular | Dünnfilmsäulen | 0,1 – 0,53 | 0,1 – 1,0 µm |
| SCOT | Support coated open tubular | Dünnschichtsäulen | ca. 0,5 | 20 – 30 µm |
| PLOT | Porous layer open tubular | mikrogepackte Säulen | 0,3 – 0,5 | 8 – 15 µm |
Wichtige stationäre Phasen zur Kapillar-GC
Die Auswahl der entsprechenden stationären Phasen erfolgt nach Polarität der zu trennenden Komponenten, Beispiele hierfür sind in der nächsten Tabelle zu finden.
Wichtige Stationäre Phasen (Kapillarsäulen) mit Anwendungsbeispielen:
| Handelsbez.: | Trägermaterial | Temp.- bereich | Polarität der Phase | Anwendungen: |
| SE-30, DB-1³, IV-1, HP-15,, RTX-16, SPB-14 | 100% Polydi- methylsiloxan | -60 bis 300/325°C | unpolar | PCB’s, Phenole, Pestizide, unpolare KW’s (petrochemische Anwendungen) |
| DB-5³, SE-54, OV-5, HP-55, RTX-56, SPB-54 | (5%)-Diphenyl-(95%)-Dimethylpoly-siloxan Copolymer | -60 bis 300/325°C | unpolar | Alkaloide, FS-Methylester, halogenierte Verbindungen |
| HP-175, SP-22504 | (50%)-Phenyl-(50%)-Methylsiloxan | 0 bis 280°C | mittel-polar | Drogen, Steroide, Pestizide, Glykole |
| HP-505, DB 17³, OV-17, RTX-506, SPB-504 | (50%)-Diphenyl-(50%) dimethylsiloxan Copolymer | 30 bis 260/280°C | mittel-polar | Steroide, Pestizide, Glykole |
| DB-1301³, HP-13015, RTX-13016 (DB-624³, RTXVolatiles6, RTX-6246 etc.)² | (6%)-Cyanopropyl-phenyl-(94%)-dimethylsiloxan Copolymer | -20 bis 260° C | mittel-polar | flüchtige halogenierte Verbindungen (EPA Methode 501, 503.1, 524.2, 601, 602, 603, etc.) |
| FFAP5 (Free Fatty Acid Phase), DB-FFAP³, CP-WAX³, STABILWAX-DA6, NUKOL | Polyethylenglykol verestert mit 2-Nitroterephthal-säure (Free Fatty Acid Phase) | 60 – 220/240°C | polar | Alkohole, Aldehyde, Carbonsäuren (FS) |
| HP PLOT MOLESIEVE5, RT-MISIEVE 13X6, SUPELCO MOLSIEVE 5A PLOT4 | Molekularsieb | bis 350° C | Gase, Permanentgase | |
| DB-502.2³, RTX-502.26, RTX-Volatiles5, HP-VOC5 | -60 bis 280° C | “purgable” organische Verbindungen (EPA Method 502.2) 524.2, 601, 602, 8024, 8260) | ||
| DB-608³, HP-6085, SPB-6084 | -30 bis 310° C | „Umweltanalyse“, chlorierte KW’s, EPA 608, 508, 8080, 8081, 8150 |
Typische Einsatzgebiete der GC und Randtechniken
Die GC ist eine sehr effiziente Trennmethode, die für die Substanzen (möglichst gelöst in Lösungsmittel) geeignet ist, welche sich möglichst unzersetzt in den gasförmigen Zustand überführen lassen. Typische Beispiele aus unserem Alltag sind Alkoholbestimmungen in Wein, Bier oder auch im Blut (Blutalkohol), Überprüfungen von ätherischen Ölen in Badezusätzen oder Medikamenten.
Auswahlkriterien
Für die richtige Auswahl von stationärer Phase, Detektor und Injektor müssen mehrere Faktoren berücksichtigt werden:
- Die Polarität der Probe ist wichtig für die richtige Auswahl der stationären Phase
- Die Substanzmenge ist wichtig für die Dimension der Säule, die Art der Detektion und ggf. für das passende Injektionssystem
- Der Aggregatzustand der Probe ist ausschlaggebend für die Auswahl des Probenaufgabesystems
Derivatisierung
Viele Substanzen, die nicht genügend flüchtig sind, können durch eine geeignete Derivatisierung flüchtig gemacht werden. Es gibt eine Vielzahl von Derivatisierungsreagenzien und alle haben unterschiedliche Einsatzgebiete. Aber einige haben sich durchgesetzt, weil sie einfach zu handhaben oder universell einsetzbar sind.
Die folgende Tabelle zeigt einige wichtige Derivatisierungsreagenzien und ihre Einsatzgebiete:
| Derivatisierungsreagenz: | Derivatisierung von: | Prinzip |
| Diazomethan, Methyliodid oder –bromid | -OH, -COOH, R-NH2 | Alkylierung |
| Acetanhydrid | -OH, R-NH2 | Acylierung |
| Silylierungsmittel (z. B. Trimethylchlorsilan) | -OH, *COOH | Silanisierung |
Randtechniken
Viele neuere Entwicklungen zeigen, dass man vor allem bei der Probenaufgabe in der GC viele Variationsmöglichkeiten hat. Davon sind in der folgenden Tabelle einige vorgestellt (diese Tabelle stellt nur einen Auszug dar):
| Probenaufgabetechnik: | Probe: | Ziel: |
| Head-Space | Flüssig/ Fest | Bestimmung von leicht flüchtigen Komponenten in Lösung z. B. ätherische Öle in Bädern oder Alkohol in Blut oder Arzneimittel |
| Pyrolyse | fest | thermische Zersetzung -> Analyse von Zersetzungsprodukten (z. B. bei Polymeren) |
| Thermo-Desorption oder Trap-Technik | flüssig oder gasförmig | Organ. Komponenten werden auf Trägern gebunden zur Vorreinigung und/oder Probenaufkonzentration und dann thermisch desorbiert (z. B. Umweltgifte, bei Raumluft-messungen u.a. chlorierte Kohlenwasserstoffe etc.) |
Die so genannte EPC (electronic pressure control) ist heute meist Standardausstattung eines GC’s und wird besonders im Hinblick auf genaue Gasflussdosierung (bessere Reproduzierbarkeit der Retentionszeiten) und Einsparung von Trägergasen auch immer mehr Verbreitung finden.
Checkliste für die GC
Was muss ich vor Beginn der Messung beachten?
Elektrische Verkabelung:
- Hat sich nichts gelockert?
Gasversorgung:
- Ist alles dicht? Überprüfen mit einem „Ieakfinder“
- Sind die installierten Sauerstoff-, Feuchtigkeits- und Kohlenwasserstofffallen OK (evtl. austauschen!)?
Gase:
- Haben die Gase die erforderliche Reinheit (mind. 99,9995% = 5.5)?
- Ist noch genügend Gas in der Flasche (Druck?)?
- Ist das Trägergas richtig eingestellt? Die Brenngase überprüfen.
- Muss ein Make-up-Gas zudosiert werden?
- Wie ist das Splitverhältnis?
- Wie ist der Septumpurge?
Probe:
- Muss die Probe derivatisiert werden? Wie ist sie gelöst?
Injektor:
- Beim manuellen Injektor sicherstellen, dass die Injektionsspritzen sauber sind, um Kontaminationen zu vermeiden, ggf. mit Aceton spülen.
- Beim automatischen Probengeber nachprüfen, ob die Waschflüssigkeiten reichen.
- Probeneinlass säubern. Bei Problemen mit Kontaminationen auch an den Liner oder die Septen denken. Liner säubern?
- Septen hochtemperaturtauglich?
Säule:
- Ist die Säule richtig eingebaut und nicht beschädigt (Lecks?) Überprüfen!
- Stimmen die vom Hersteller vorgegebenen Abstände im Injektor und Detektor?
- Sind die Enden der Säule richtig gekürzt worden? Gerade?
- Wurden die richtigen Schneidringe (Ferrules) eingesetzt (temperaturstabil und dicht? Graphit oder Vespel?)
Detektor:
- Brenngase und Make-up-Gase eingestellt?
- Einige Detektoren messen nicht nachweisempfindlich, weil sie entweder verschmutzt sind oder keine Make-up-Gaszufuhr haben. Nachprüfen und evtl. Detektor säubern (Düsen u. Dichtungen erneuern?)
Der richtige Säuleneinbau:
- Das richtige Installationswerkzeug besorgen: GC-Handbuch, Säulenschneider, (z. B. aus Keramik), Säulenverschraubungen und Schneidringe, Lupe und temperaturstabile Handschuhe.
- Für Installation im Injektor und Detektor sollten von der Säule etwa 50 cm auf jeder Seite abgewickelt werden.
- Säule auf jeder Seite etwa 4 – 5 cm abschneiden (z. B. mit Keramikschneider). Auf geraden „Abbruch“ achten! Nachdem man mit dem Keramikschneider die Säule angeritzt hat legt man die Säule so zwischen Daumen und Zeigefinger, dass die Schnittstelle genau zwischen Daumen und Zeigefinger liegt. Man versucht dann beide Finger sehr nahe zusammen zu bringen und versucht leicht zu ziehen und gleichzeitig zu biegen. Sollte die Säule dann nicht auseinander brechen, muss man eine andere Stelle anschneiden und es noch einmal versuchen. Keine Gewalt anwenden, da dann die Säulen unkontrolliert brechen.
- Säulenenden mit einer Lupe betrachten und darauf achten, dass die Kanten ganz gerade sind. Keine Glas- oder Polyimidstücke dürfen an den Enden hängen.
- Säule in den Kapillarsäulenhalter im Ofen hängen.
- Die Verschraubung und die Schneidringe sollten etwa 5 – 10 cm auf die Säule geschoben werden (Richtung beachten).
- Die Säule zuerst in den Einlass installieren. Im Handbuch nachsehen um die richtige Länge in den Einlass einzuführen. Die Säule dementsprechend in den Einlass einführen ggf. markieren (z. B. ein Septum an die Stelle schieben). Die Verschraubung und den Schneidring nach oben schieben und an der markierten Stelle mit der Hand eindrehen. Dann mit dem Schraubenschlüssel noch ¼ bis eine ½ Umdrehung festziehen. Achtung: Keine Gewalt!
- Trägergas anstellen und einen genügend hohen Fluss einstellen (0,5 – 2 ml/min). Am Ende ein Gläschen mit Aceton stellen, die Säule hineinhalten und schauen ob Bläschen entstehen.
- Die Säule in den Detektor installieren. Wieder im Handbuch nach den richtigen Abständen nachsehen.
- Nach Lecks überprüfen. Nie eine Säule hochheizen ohne vorher nach Lecks geprüft zu haben. Die Säule eine Weile nur mit Trägergas spülen lassen.
- Erst jetzt die Temperaturen vom Injektor und Detektor einstellen.
- MAKE UP und Detektorgase einstellen.
Wie fange ich die Arbeit mit einem GC-Gerät an?
- Zuerst checken, in welchem Zustand sich das System befindet. (Siehe Checkliste)
- Falls noch nicht vorhanden die Säule einbauen. Wichtig sind hier nicht nur die richtigen Abstände (Herstelleranweisungen befolgen), sondern auch die richtige Technik beim Kürzen. (Siehe Säuleninstallation)
- Wie sind die eingestellten Trägergasflüsse? An dieser Stelle sollte man mit einem Blasenflussmesser die Gasflüsse kontrollieren. Wenn die Säule eingebaut ist, sorgen Sie dafür, dass genügend Fluss über die Säule geht. Kapillarsäulen ca. 1 ml/min.
- Wenn Sie eine Säule eingebaut haben, dann erkundigen Sie sich erst über die Temperaturlimits und das Packungsmaterial und stellen erst dann die Temperaturen am Injektor und Detektor ein.
Cave: Nie Säulen heizen, wenn kein Fluss über die Säule geht!
Um die Säule zu reinigen sollten Sie diese erst einmal ausheizen. - Die Gase für den Detektor einstellen (MAKE UP-Gas, Brenngase etc.). Der FID braucht Wasserstoff und synthetische Luft. Richtige Einstellung der Gase hängt von der Bauart ab. Im Handbuch nachlesen.
- Die Säule sollte ca. 20° C über der maximalen Temperatur der Proben eingestellt und ca. 2 Stunden vorkonditioniert werden. Sollte die Basislinie nicht ein bisschen abfallen kann man die Säule bis zur maximalen Temperatur der Säule ausheizen. Wenn die Basislinie immer noch nicht nach 10 min. abfällt, muss man noch mal auf Lecks überprüfen.
- Die Temperaturen für die Analytik einstellen. Der Detektor sollte immer über 100° C betrieben werden (besser ca. 180° C), der Injektor sollte die richtige Temperatur für die Proben haben (niedrig oder hoch siedende Verbindungen?), aber auch möglichst weit über 100° C eingestellt werden, damit die Proben gleichmäßig verdampfen könnten.
- Sich Zeit lassen, damit die Säule, Injektor und die Detektoren warm werden. In der Zwischenzeit die Proben vorbereiten.
- Wenn Sie keine Vergleichsproben in Ihrem Labor haben, können Sie folgende nicht retendierende Substanzen injizieren: Butan und/oder Methan für den FID und Splitinjektor. Headspace Gase von Acetonitril (NPD), Methylenchlorid (ECD), Luft (TCD/WLD), Argon (MSD). Eine gute Installation ist durchIst alles in Ordnung, können Sie jetzt einen Standard injizieren. Stimmt das sich ergebende Chromatogramm mit einem Vergleichschromatogramm überein, stimmen Peakhöhe und Retentionszeit, so ist Ihr System in Ordnung und Sie können mit Ihren Messungen beginnen. symmetrische Peaks zu erkennen.
- Ist alles in Ordnung, können Sie jetzt einen Standard injizieren. Stimmt das sich ergebende Chromatogramm mit einem Vergleichschromatogramm überein, stimmen Peakhöhe und Retentionszeit, so ist Ihr System in Ordnung und Sie können mit Ihren Messungen beginnen.
- Möchten Sie Ihre Arbeit für heute beenden, am nächsten Tag aber weiterarbeiten, lassen Sie das Trägergas bei niedrigem Fluss, ca. 0,2 – 0,5 ml/min über die Säule laufen. Wenn Sie sicher sind, dass über Nacht nicht das Trägergas ausgeht, dann können Sie auch den Ofen noch heizen lassen ansonsten den Ofen ausstellen. Die Brenngase oder Make-up-Gase können Sie über Nacht auch ausstellen, ist aber bei manchen Detektoren besser sie anzulassen (z. B. NPD).
- Möchten Sie das System längere Zeit nicht verwenden, heizen Sie die Säule erst aus und bauen sie nach dem Abkühlen aus. Dann erst sollten Sie das gesamte Gerät abkühlen und wenn es abgekühlt ist ausstellen.
Von IUPAC empfohlene Symbole für die Chromatographie
Im Jahre 1993 erschienen in Pure & Appl. Chem., Vol 65, No. 4, pp. 819-872 Empfehlungen von der IUPAC (International Union of Pure and Applied Chemistry) zur „Nomenklatur für die Chromatographie“. Der Artikel über 53 Seiten ist ebenfalls im „ChromBook“ der Fa. Merck, Darmstadt, nachzulesen. Dieser enthält Definitionen, Symbole, Begriffe und Erläuterungen zur Chromatographie. Nachfolgend sind die wichtigsten Größen mit dem entsprechenden Symbol zusammengestellt.
| Größe | englische Bezeichnung | Symbol |
| Trennfaktor | separation factor | α |
| (Bis 1993: Selektivitätsfaktor | selectivity factor | α) |
| Fläche | area | A |
| Durchmesser | diameter | d |
| Diffussionskoeffizient | diffusion coefficient | D |
| Flussrate | flow rate (volumetric) | F |
| Bodenhöhe | plate height | H |
| Viskosität | viscosity | η |
| Gleichgewichts (Verteilungs-) konstante (Koeffizient) | equilibrium (distribution) constant | K |
| Retentionsfaktor | retention factor | k |
| (Bis 1993: Kapazitätsfaktor | capacity factor | k’) |
| Länge | length | L |
| Bodenzahl | plate number | N |
| Dichte | density | ρ |
| Druck | pressure | p |
| relativer Druck | pressure, relative | P |
| Radius | radius | r |
| Temperatur | temperature (absolute) | T |
| Zeit | time | t |
| lineare Geschwindigkeit | velocity (linear) | u |
| (Retentions) Volumen | volume | V |
| Masse (Gewicht) | mass (weight) | W |
| Peakbreite | peak width | w |
Gebräuchliche Abkürzungen
| Abkürzung: | Engl. Bezeichnung | Deutsche Übersetzung |
| ECD | electron capture detector | Elektroneneinfangdetektor |
| EPC | electronic pressure control | Elektronische Druckkontrolle |
| FID | flame ionisation detector | Flammenionisationsdetektor |
| GC | gas chromatography | Gaschromatographie |
| GC-MS | gas chromatography / mass spectrometry | Gaschromatographie-Massenspektroskopie |
| GLC | gas liquid chromatography | Gasflüssigkeitschromatographie |
| GSC | gas-solid-chromatography | Gasfestchromatographie |
| HETP | height equivalent to a theoretical plate | Theoretische Trennstufenhöhe |
| MS | mass spectrometry | Massenspektrometrie |
| NPD | nitrogen phosphorous detector | Stickstoff-Phosphor-Detektor |
| PLOT | Porous layer open tubular | mikrogepackte Säule |
| PTV | programmed temperature vaporizer | Temperaturprogrammierte Verdampfung |
| RT | retention time | Retentionszeit |
| SCOT | Support coated open tubular | Dünnschichtsäulen |
| SFC | supercritical fluid extraction | Überkritische Flüssigkeitsextraktion |
| TCD | thermal conductivity detector | Wärmeleitfähigkeitsdetektor |
| WCOT | Wall coated open tubular | Dünnfilmsäulen |
| WLD | thermal conductivity detector | Wärmeleitfähigkeitsdetektor |
Einige GC-Hersteller
| Firma | URL | Bemerkungen |
| Agilent Technologies | www.agilent.com | Ehemals Hewlett-Packard, J&W oder Varian (Chrompack) |
| Thermo Fisher Scientific | www.thermofisher.com | u.a. ehemals Carlo Erba |
| Perkin Elmer | www.perkinelmer.de | |
| Shimadzu | www.shimadzu.de | |
| Supelco | www.sigmaaldrich.com | Nur Zubehör (u.a. Säulen) |
| Restek | www.restek.com | Nur Zubehör (u.a. Säulen) |
| Phenomenex | www.phenomenex.com | Nur Zubehör (u.a. Säulen) |
| Gerstel | www.gerstel.de | u.a. Sonderanfertigungen |
Literatur
Ältere Literatur, „GC-Klassiker“
- Baars B., Schaller H., Fehlersuche in der Gaschromatographie, Weinheim: VCH (1994)
- Ettre L. S., Hinshaww J. V., Rohrschneider L., Grundbegriffe und Gleichungen der Gaschromatographie, Heidelberg: Hüthig Verlag (1996)
- Grob K., Marking and Manipulating Capillary Columns for Gas Chromatography, Heidelberg: Hüthig Verlag (1986)
- Kolb, B. Gaschromatographie in Bildern: Eine Einführung, Weinheim, Wiley-VCH (2003)
- Engewald, W.,Struppe, H.G. Gaschromatographie, Berlin, Springer (2000)
- Knapp D. R., Handbook of Analytical Derivatization Reactions, New York: John Wiley & Sons (1979)
- Nikelly J. G., Advances in Capillary Chromatography, Heidelberg: Hüthig Verlag (1985)
- Oehme M. Praktische Einführung in die GC/MS-Analytik mit Quadrupolen, Heidelberg: Hüthig Verlag (1996)
- Schomburg G., Gaschromatographie, Weinheim: VCH (1987)
- Schwedt G., Chromatographische Trennmethoden, Stuttgart: Georg-Thieme Verlag (1979)
Neuere Literatur (seit 2010)
… und in Englisch:
- David O. Sparkman, Gas Chromatography and Mass Spectrometry: A Practical Guide, Academic Press, 2. Auflage (2011)
- Colin Poole, Gas Chromatography, Elsevier Ltd, Oxford (2012)
- Carol Evans, Encyclopedia of Gas Chromatography: Volume 2-4, Ny Research Press (2015)
- Katja Dettmer-Wilde, Werner Engewald (Hrsg.) Practical Gas Chromatography: A Comprehensive Reference, Springer (2014)
- Austin V. Signeur, Guide to Gas Chromatography Literature, Springer (2012)
- Henry J. Noebels, R.F. Wall, Gas Chromatography, Ulan Press (2012)
- Raymond P W Scott, Gas Chromatography, The Reese-Scott Partnership (2012)
- Raymond P.W. Scott, Gas Chromatography Detectors, Integritext UK (2015)
- Hans-Joachim Hübschmann, Handbook of GC/MS, 3. Auflage, Wiley VCH (2015)
- E-Book: Advancing GC Methods and Environmental Analysis, kann nach Anmeldung kostenlos bei LCGC, UBM, downgeloaded werden (2016)
Internet
Im Internet findet sich zum Teil sehr gutes Text- und Anschauungsmaterial (Videos, Animationen etc.) zur GC. Aus der Fülle an Homepages seien nur folgende drei genannt, diese enthalten umfangreiche und gute Informationen:

