Startseite » Validierung » Nützliches » Kompakte Informationen »
Fließschema zur Methodenvalidierung einer „Spurenmethode“ mittels Chromatographie
Literaturliste zum Thema (Auswahl)
Was versteht man unter Validierung?
Begriffsbeschreibung Validierung
Unter Validierung versteht man den Nachweis und die Dokumentation der Zuverlässigkeit einer Methode. Die Definition nach DIN ISO 9000 lautet: Bestätigen aufgrund einer Untersuchung und durch Bereitstellung eines objektiven Nachweises, dass die besonderen Forderungen für einen speziellen beabsichtigten Gebrauch erfüllt worden sind. Diese Definition ist sehr allgemein gehalten und lässt dem Fachmann freie Entscheidungsräume. Leider wird zunehmend seitens der Inspektoren und der Behörde bei Inspektionen oder Anmeldungen eine Liste mit Maximalforderungen stur „abgehakt“. Mit der Validierung beschäftigt man sich intensiver seit dem verstärkten Einzug von QS-Systemen in die Laboratorien.
Wer fordert Validierung
Besonders die Einführung der Akkreditierung nach EN 45001 und die Forderungen der amerikanischen Gesundheits- und Umweltbehörden (FDA, EPA), sowie die DIN EN ISO/IEC 17025: 2018 sind als Anstoß zu bewerten. Im Folgenden werden die Elemente der Validierung vorgestellt und eine pragmatische Umsetzung im analytischen Labor vorgestellt.
Validierung und Analytik
Es fehlt eine verbindliche Definition des Begriffes Validierung speziell für die Analytik, auch der Umfang wird unterschiedlich festgelegt. Bei Sucker finden sich sieben allgemeine Validierungsthesen. Für die Analytik abgewandelt, könnten diese Thesen wie folgt lauten:
Validierungsthesen für die Analytik
- Validierung ist ein Arbeitsinstrument zur Qualitätssicherung neben anderen wie SPC (Statistical Process Control)
- Validierung ist produkt- und zweckspezifisch auszuführen. Die Verantwortung über Ausmaß und Art liegt beim Analytiker
- Validierung heißt, das Notwendige tun, um eine Eskalation zu vermeiden. Alle kritischen Schritte müssen validiert werden, aber nicht wahl- und kritiklos alles
- Methodenvalidierung beginnt am Besten beim Endergebnis und geht im Analysenablauf bis zum ersten Schritt zurück
- Methodenvalidierung beginnt am Besten beim Endergebnis und geht im Analysenablauf bis zum ersten Schritt zurück
- Validierung kann nicht durch Abhaken von Resultaten mittels Checkliste erfolgen
- Nach Möglichkeit ist die statistische Relevanz und damit die Messunsicherheit zu ermitteln. Eine fehlerhafte Analytik („wahrer“ Wert) gibt es nicht
- Für Ergebnisse aus validierten Methoden sind Art und Häufigkeit der notwendigen Kontrollen festzulegen mit dem Ziel, den Gesamtanalysenaufwand zu minimieren, aber dennoch die erforderliche Ergebnissicherheit zu erzielen
Statistische Daten
Im Rahmen der Validierung werden statistische Daten ermittelt. Oft muss der Anwender entscheiden, ob ein Wert nun ein Ausreißer ist oder nicht. Eine nicht zu empfehlende Praxis ist die subjektive Beurteilung.
Ausreißertests
Nicht nur in einer Abteilung, sondern in ganzen Bereichen müssen objektive Kriterien zu einer Ja/Nein-Entscheidung festgelegt und zwingend befolgt werden (z. B. mittels Dixon, Grubbs-Test oder 20% Abweichung vom Mittelwert). Sonst ist eine wichtige Voraussetzung für die Vergleichbarkeit von Ergebnissen nicht erfüllt.
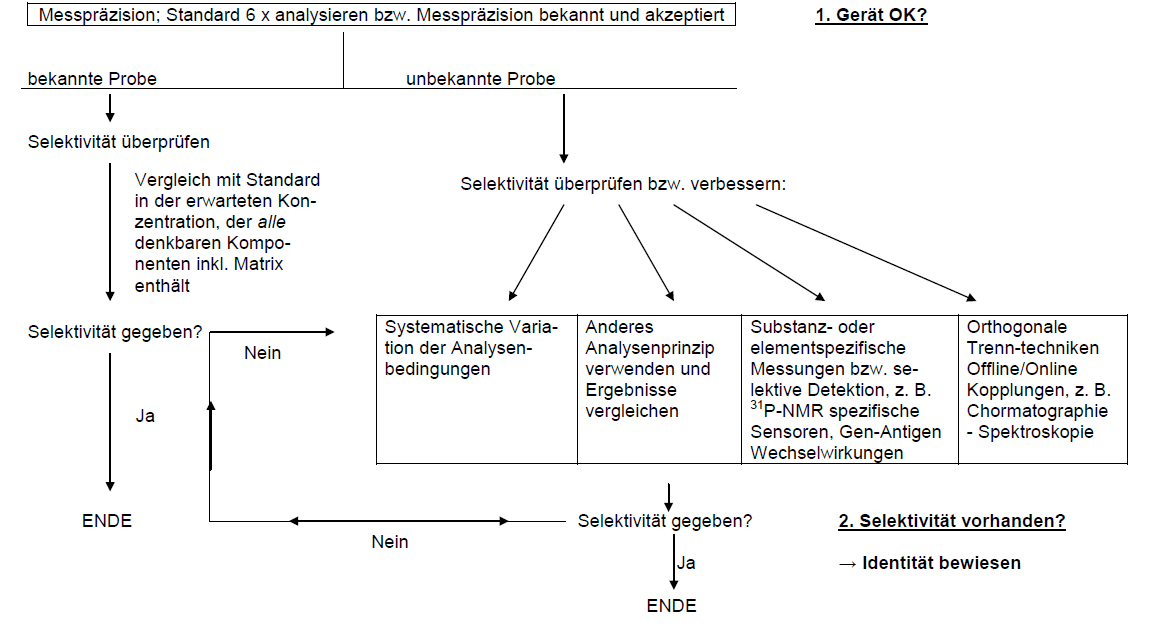
Überprüfung des Messgeräts
Eine gute Methode kann nur in einem „guten“ Gerät „gute“ Ergebnisse liefern. Die Messpräzision, d. h. die Güte der verwendeten Apparatur muss bekannt sein. Diese kann durch entsprechende Gerätetests ermittelt werden.
Prüfmittelüberwachung
Die im Rahmen der Prüfmittelüberwachung durchzuführende Kalibrierung kann bei einfachen Geräten gegebenenfalls die aufwendigeren Gerätetests ersetzen. Auf die Gerätetests wird hier nicht näher eingegangen (siehe Dokument ALC 00297). In der Zwischenzeit bieten mehrere Hersteller entsprechende Softwaremodule an, welche die Durchführung dieser Tests erleichtern.
Verfahrensvalidierung
Die Einbeziehung aller relevanten Einflüsse auf das Ergebnis (Probevorbereitung, Messung, Messgerät, Datengenerierung) wird durch den Begriff Verfahrensvalidierung unterstrichen. Doch scheint sich in der Analytik (vorerst) der Begriff „Methodenvalidierung“ durchgesetzt zu haben. Dennoch: Immer häufiger ist in jüngster Zeit von „Verfahren“ („process“) die Rede, siehe weiter unten „Trends auf dem Gebiet der Validierung“.
Kritische Schritte einer Validierung
Bei der Validierung sollten gerade die kritischen Schritte der Methode überprüft werden. Wenn möglich und sinnvoll, sollte in besonderen Fällen die Probenahme in die Methodenvalidierung aufgenommen werden. Eine nicht repräsentative Probe kann das Ergebnis einer sonst hervorragenden Methode zunichte machen. Hat der Anwender im Labor keinen Einfluss auf die Probenahme, so sollten diese und eventuell auch der Probetransport sowie die Lagerung genau beschrieben und dokumentiert werden.
Folgende Voraussetzungen gelten für die Methodenvalidierung:
- Der Zweck ist unmissverständlich definiert
- Die Validierung erfolgt unter realen und nicht unter optimalen Bedingungen
- Es liegt eine ausgereifte, bereits optimierte Methode schriftlich vor. Diese Forderung ist nicht immer realisierbar. In der Praxis sind oft Methodenentwicklung und einzelne Schritte der Validierung (Selektivität, Linearität, Robustheit) miteinander verknüpft
- Das Gerät hat eine bekannte und akzeptierte Präzision (Messpräzision)
- Das Personal ist mit der Methode vertraut
- Die verwendeten Chemikalien (chromatographische Säulen, Referenzsubstanzen, Lösungsmittel, Reagenzien etc.) sind von guter (und bekannter) Qualität
Validierungselemente und deren Überprüfung
Maximalumfang einer Validierung
Mittels der Wiederfindungsrate wird überprüft, ob bei der Probeaufarbeitung (z. B. Extraktion, Derivatisierung, Injektion) möglicherweise ein Teil der Substanz „verschwindet“.
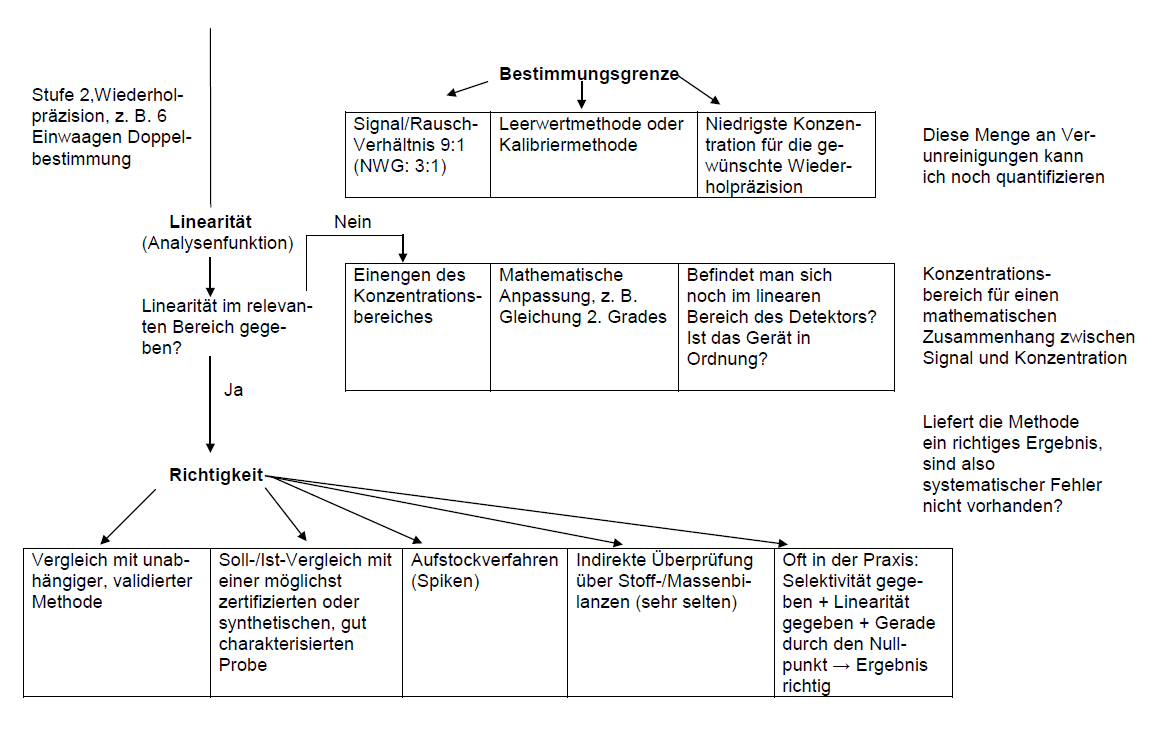
Richtigkeit
Richtigkeit
Die Richtigkeit ist ein Maß für die Abweichung des Messwertes vom richtigen Wert (manchmal als „wahrer“ Wert bezeichnet) aufgrund eines systematischen Fehlers.
Das Fehlen von systematischen Fehlern ist somit eine Grundvoraussetzung für die Richtigkeit. Weitere Voraussetzungen sind:
- Die Methode ist selektiv.
- Die Wiederfindungsrate beträgt nach jedem Schritt der Probenvorbereitung 100 % oder ist konstant oder bekannt und rechnerisch korrigierbar.
Prüfung auf Richtigkeit
- Vergleich mit einem Referenz- oder Arbeitsstandard (Soll/Ist-Vergleich)
- Vergleich mit einer unabhängigen, möglichst validierten Methode.
- Aufstocken („Spiken“ einer Probe)
Wenn bei bestimmten Proben (z. B. Wirkstoffe) keine der drei Methoden anwendbar ist, kann als Kriterium für die Richtigkeit folgendes gelten:
Die Selektivität ist erwiesen, Linearität ist vorhanden, und die Kalibriergerade geht durch den Nullpunkt
Selektivität
Oft werden die Begriffe Spezifität und Selektivität für den gleichen Sachverhalt verwendet, daher sei hier die korrekte Definition der beiden Begriffe aufgeführt:
Spezifität
- Eine Methode arbeitet selektiv, wenn sie verschiedene, nebeneinander zu bestimmenden Komponenten ohne gegenseitige Störungen erfasst
Prüfung auf Selektivität
Prüfung auf Selektivität:
- Prüfung auf Richtigkeit: Da Selektivität eine der Voraussetzungen für Richtigkeit ist, ist eine richtige Methode automatisch auch selektiv
- Vergleich mit einem Standard, der alle denkbaren Komponenten inkl. Matrix erhält
- Systematische Variation der Analysen- bzw. Messbedingungen
- Vergleich mit dem Ergebnis nach einem anderen Analysenprinzip
Selektivität in der Chromatographie
Spezialfall Chromatographie
- Ein pragmatischer (und gleichzeitig der sicherste!) Weg ist der Vergleich des erhaltenen Chromatogramms mit einem „Muster“-Chromatogramm, das sämtliche Nebenkomponenten, Verunreinigungen etc. enthält.
Vergleichschromatogramm
Die Aussage zur Selektivität kann durch den Vergleich von chromatographischen Kenngrößen unterstützt werden (relative Retention, Peakbreite, Asymmetriefaktor)
Spiken
- Sukzessive Zugabe der einzelnen Analyten und Überprüfung der chromatographischen Auflösung
- Wechsel der Säule/DC-Platte und/oder der mobilen Phase
Kopplung
- Erhöhung der Peakkapazität, z. B. durch Online-Kopplung, verschiedener chromatographischer Verfahren (LC-GC, LC-DC, SFC-GC) oder im Offline-Modus: „Schneiden“ des Peaks oder einer Peakfraktion und Untersuchen der Fraktionen mit anderen Trennmethoden und/oder Spektroskopie
Ratio Plot
- Ratio-Plot: Detektion bei zwei Wellenlängen, Prüfung der Konstanz der Extinktionsverhältnisse (nicht sicher)
Spektren
- Online-Spektrenaufnahmen und -vergleich (UV, MS) in aufsteigender/abfallender Peakflanke und im Maximum (ggf. Spektrendatenbank) (üblich und gut, nicht 100% sicher)
Peakformenvergleich
- Peakformvergleich des Analyten in der Kalibrierlösung und der Probe (Peakbreite, Asymmetrie, Ableitungen)
Wiederfindungsrate
Wiederfindungsrate
Mittels der Wiederfindungsrate wird überprüft, ob bei der Probeaufarbeitung (z. B. Extraktion, Derivatisierung, Injektion) möglicherweise ein Teil der Substanz „verschwindet“.
Vorschlag zur Überprüfung der Wiederfindungsrate
Überprüfung der Wiederfindungsrate
Es werden insgesamt drei Lösungen hergestellt und analysiert:
Lösung 1: zu V1 ml Probenlösung V2 ml Kalibrierlösung geben, man erhält Signal S1
Lösung 2: zu V1 Probenlösung V2 Lösungsmittel geben, man erhält Signal S2
Lösung 3: zu V1 Lösungsmittel V2 Kalibrierlösung geben, man erhält Signal S3
Die Wiederfindungsrate W errechnet sich wie folgt:
W = (S1-S2)/S3 x 100
Präzision
Man unterscheidet zwischen Systempräzision (Messpräzision) und Methodenpräzision.
Messpräzision
- Die Messpräzision ist ein Maß für die Schwankungen, die durch das Analysengerät selbst verursacht werden. Sie wird durch die Mehrfachanalyse (z. B. sechsfach) eines Standards ermittelt. Die Forderung an die Messpräzision hängt vom Analysengerät ab. Bei der HPLC und GC sollte der Variationskoeffizient (Quotient aus Standardabweichung und Mittelwert) VK kleiner ca. 1% sein.
- Die Methodenpräzision beschreibt die zufällige Streuung der Analysenergebnisse. Sie wird durch eine mehrfache (meist sechsfache) Durchführung der gesamten Analyse, d. h. vom Abwiegen über die Probevorbereitung bis zu der Messung und Befundung ermittelt (sechs Einwaagen realer Proben).
Wiederholpräzision
Es wird zwischen Präzision unter Wiederholbedingungen (Wiederholpräzision, Wiederholbarkeit: Ein Labor, ein Gerät, ein Prüfer) und Präzision unter Vergleichsbedingungen (Vergleichspräzision, Vergleichbarkeit, Übertragbarkeit, Reproduzierbarkeit: Mehrere Labors, mehrere Prüfer, mehrere Geräte) unterschieden.
Laborpräzision
Das ist die Präzision innerhalb eines Labors, wenn die Bestimmung von verschiedenen Personen an verschiedenen Geräten und an unterschiedlichen Tagen evtl. mit unterschiedlichen Chemikalien etc. durchgeführt wird.
Akzeptankriterien
| Das ist die Präzision innerhalb eines Labors, wenn die Bestimmung von verschiedenen Personen an verschiedenen Geräten und an unterschiedlichen Tagen evtl. mit unterschiedlichen Chemikalien etc. durchgeführt wird. |
| Die Akzeptanzkriterien hängen stark von den Forderungen bei der speziellen Fragestellung ab. Wird beispielsweise im Pharmabereich in der Regel für die Vergleichspräzision ein VK < 2% verlangt, so sind in der Umweltpolitik VK-Werte von ca. 5-10% und in der Medizin von 10-20% durchaus akzeptabel. |
Genauigkeit
Genauigkeit
Die Genauigkeit ist kein Validierungselement, sondern der Oberbegriff für Richtigkeit und Präzision. Ein Ergebnis ist genau,
wenn es frei von zufälligen (Ergebnis ist präzise) und systematischen Fehlern (Ergebnis ist richtig) ist. Angemerkt sei an dieser Stelle, dass in der Literatur die Begriffe „Genauigkeit“ und „Richtigkeit“ oft als synonyme Terme verwendet werden.
Linearität
Kalibrierfunktionen
Eine Methode ist in einem bestimmten Konzentrationsbereich linear, wenn das Messsignal direkt proportional zu der Analytkonzentration in der Probe ist (nicht im Standard!). „Direkt proportional“ bedeutet nicht zwingend eine lineare Abhängigkeit zwischen Messsignal und Analytkonzentration! Aus diesem Grunde mag der Begriff „Analysenfunktion“ oder Kalibrierfunktion treffender sein. Kalibrierfunktionen zweiten Grades können und sollten bei Bedarf verwendet werden.
Linearität des Gerätes
Ähnlich der Messpräzision kann hier mit einer Standardlösung die Linearität des Gerätes bzw. des Detektors bestimmt werden. Die Linearität der Methode ist meist kleiner (selten gleich) als die Linearität des Detektorsystems. Gleichheit bedeutet, dass die Matrix und die Probevorbereitung keine systematischen Fehler verursachen.
Prüfung der Methode
Prüfung der Linearität:
Üblicherweise wird das Signal S gegen die Konzentration c aufgetragen. Die Steigerung dS/dc ist ein Maß für die Empfindlichkeit der Methode. Wenn die Steigerung dS/dc konstant (linearer Bereich) und das Signal s bei c = 0 ebenfalls gleich Null ist, ist eine Einpunktkalibrierung zulässig.
Einpunkt-Kalibrierung
Wenn die Steigerung dS/dc konstant (linearer Bereich) und das Signal s bei c = 0 ebenfalls gleich Null ist, ist eine Einpunktkalibrierung zulässig. Ansonsten sollten mindestens fünf Konzentrationen vermessen werden, um den mathematischen Zusammenhang zwischen Masse und Signal (Regressionsmodell) genau ermitteln zu können.
Auftragung S/c gegen c
Es ist nicht immer zweckmäßig, eine Gerade durch den Nullpunkt zu zwingen. Neben der klassischen Auftragung, Signal gegen Konzentration (S/c), hat sich die Auftragung des Quotienten aus Signal und Konzentration gegen die Konzentration c bewährt; Verdünnungsfehler im unteren Bereich werden einfach erkannt.
Arbeitsbereich
Im Zusammenhang mit der Linearität wird oft der Arbeitsbereich „range“ genannt. Dieser ist der Bereich zwischen der niedrigsten und der höchsten Konzentration (Menge) des Analyten in der Probe, für den die geforderte Präzision und Genauigkeit bewiesen wurden.
Nachweis- und Bestimmungsgrenze
Bestimmungsgrenze
Die Nachweisgrenze ist die kleinste Konzentration (Menge) des Analyten in der Probe, die qualitativ noch erfasst werden kann (Ja/Nein-Entscheidung). Die Bestimmungsgrenze ist die kleinste Konzentration (Menge) des Analyten in der Probe, die mit gegebener Präzision und Richtigkeit quantitativ bestimmt werden kann. Das zugrunde liegende mathematische Modell und die Bestimmungsmethoden sind in der DIN 32645 beschrieben.
Erfassungsgrenze
Sie gibt die Konzentration (Menge) an, die mit einer Wahrscheinlichkeit von 50 % nachgewiesen werden kann. Somit kann die Erfassungsgrenze vereinfacht als die doppelte Nachweisgrenze angesehen werden.
Überprüfung der Grenzen
Überprüfung der Nachweis- und Bestimmungsgrenze:
Mit Hilfe von Makros in käuflichen (z. B. Excel) oder selbst geschriebenen Softwareprogrammen ist die Überprüfung in Anlehnung an die DIN-Norm 32645 möglich.
Vereinbarungen über die Grenzen
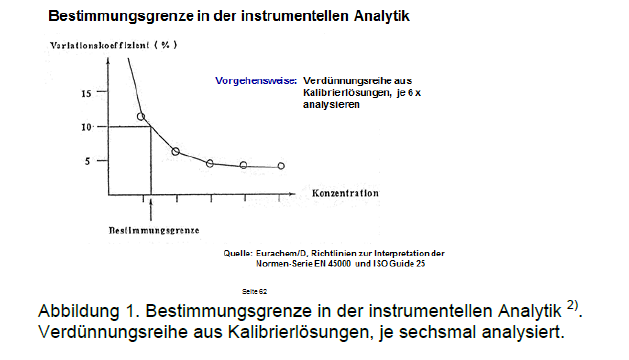
In der chromatographischen Praxis gelten zumeist das zwei- bis dreifache Rauschen als Nachweisgrenze und das neun- bis zehnfache Rauschen als Bestimmungsgrenze. Selbstverständlich entsprechen die ermittelten Werte einer Momentaufnahme; sie geben den aktuellen Gerätezustand wieder (Lampe, Güte der eingesetzten Chemikalien, etc.). Nach jedem Wechsel im System sollte die Bestimmung wiederholt werden. Ein pragmatischer Vorschlag zur Ermittlung der Bestimmungsgrenze stammt von Eurachem/D2) (Abbildung 1). Durch eine Verdünnungsreihe wird die kleinste Konzentration ermittelt, für die der Variationskoeffizient noch tolerierbar ist.
Bestimmungsgrenze in der instrumentellen Analytik
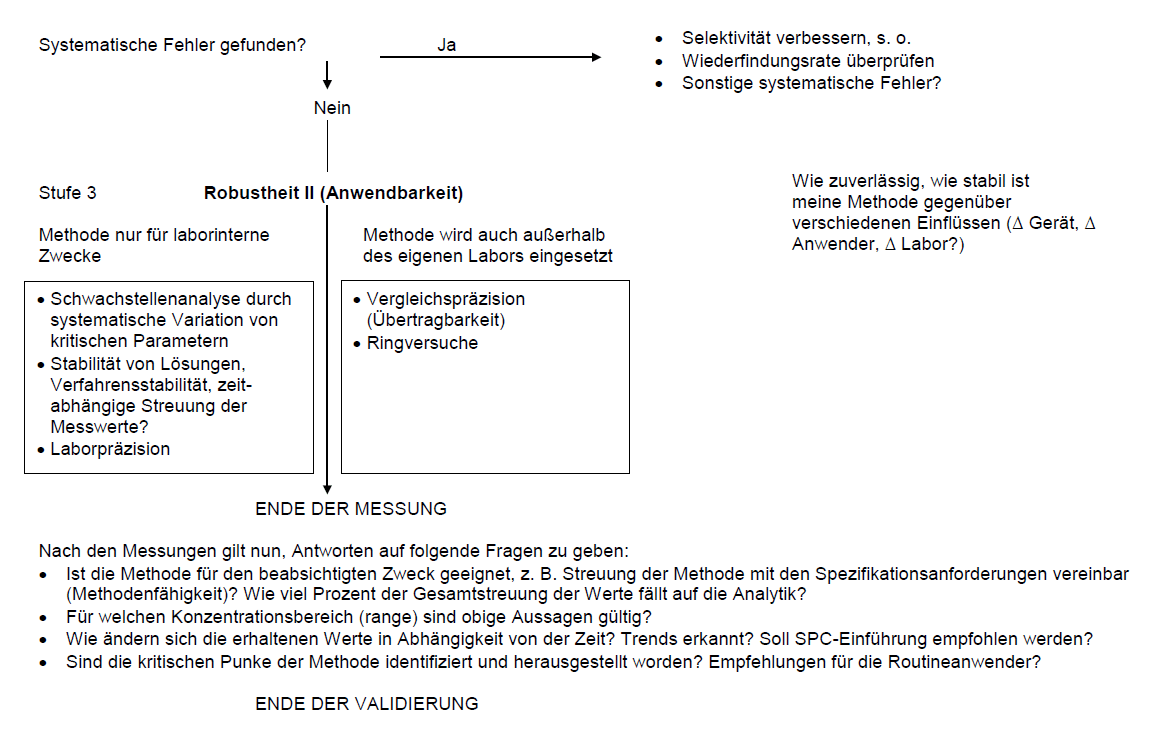
Robustheit
Überprüfung der Robustheit
Eine Methode ist robust, wenn durch (ungewollte) Änderung der Testbedingungen das Endergebnis nicht, oder nur unwesentlich verfälscht wird. Als Maß für die Robustheit wird der Bereich genannt, in dem das Ergebnis von der Änderung eines oder mehrerer Parameter unabhängig ist, z. B. „Messergebnis konstant zwischen 29° C und 31° C, pH 4,9 bis 5,1, gemessen an den Geräten A, B und C“.
- Vergleich der Messergebnisse zu Beginn und am Ende einer Analysenserie (Verfahrensstabilität).
- Vergleich von Messergebnissen laborintern (Laborpräzision) und zwischen unterschiedlichen Labors (Vergleichbarkeit, Übertragbarkeit). Die Weiterführung dieses Gedankens, nämlich der Überprüfbarkeit als Kriterium der Robustheit, führt zu den Ringversuchen (ab ca. 30 bis 40 Labors).
- Systematische Variation der Einflussparameter in einem relevanten (!) Bereich.
Umfang der Methodenvalidierung
Warum Validierung?
Die Methodenvalidierung ist eine notwendige, qualitätssichernde Maßnahme, sie muss allerdings effektiv durchzuführen sein und somit bezahlbar bleiben. Es gilt folgender Grundsatz: Der Aufwand und der Umfang der Validierung sollten in einem angemessenen Verhältnis zu den Forderungen stehen. Deswegen ist es wichtig, sich über Art und Ziel der Analytik im Klaren zu sein. So kann beispielsweise – wenn der Aufwand vertretbar ist – durch den Vergleich mit einer unabhängigen Methode die Richtigkeit der zu validierenden Methode belegt werden. Die Validierung wäre bereits damit erfolgreich beendet (Ergebnisvalidierung). Ein (aufwendiger) Ringversuch wiederum ermöglicht die Beurteilung einer Methode; hiermit werden die Präzision sowie die Robustheit des gesamten Verfahrens überprüft.
In der pharmazeutischen Analytik kann man grob folgende Methodentypen unterscheiden: Identitätstests, Gehaltsbestimmung, quantitative Spurenmethode (ICH-Richtlinien). Reicht beispielsweise im ersten Fall eine – allerdings sehr gründliche – Überprüfung der Selektivität aus, wären bei der quantitativen Bestimmung von z. B. Neben- oder Abbauprodukten sämtliche Validierungselemente zu überprüfen.
Grundbegriffe der Methodenvalidierung (Auswahl, Definitionen)
| Bezeichnung | englische Bezeichnung | Aussage über: |
|---|---|---|
| Genauigkeit | accuracy | systematische und zufällige Fehler Die Genauigkeit ist der Oberbegriff für Richtigkeit und Präzision. |
| Richtigkeit | trueness, accuracy of the mean | systematische Fehler Die Richtigkeit ist das Maß für die Abweichung vom richtigen Wert (manchmal als „wahrer“ Wert bezeichnet) aufgrund eines systematischen Fehlers belegt werden kann die Richtigkeit über die Wiederfindungsrate, internen Kontrollproben, ein zweites Verfahren oder zertifizierte Referenzproben |
| Präzision | precision | zufällige Fehler Die Präzision ist ein Maß für die Streuung der Analysewerte. |
| Wiederhol- präzision | repeatability | (Wiederholbedingungen: Ein Labor, ein Prüfer, ein Gerät) |
| Laborpräzision | intermediate precision | Ein Labor, zwei Prüfer, zwei Geräte, zwei Tage usw. |
| Vergleichspräzision | reproducibility | Präzision im Vergleich „Labor zu Labor“ (Vergleichbedingungen: Mehrere Labors, mehrere Prüfer, mehrere Geräte, also: Unterschiedliche Rahmenbedingungen) |
| Linearität | linearity | Zusammenhang zwischen Signal und Konzentration Das Messsignal muss proportional zu der Analytkonzentration in der Probe (nicht im Standard!) sein, wobei eine lineare Abhängigkeit nicht zwingend ist. Zur Prüfung der Linearität wird üblicherweise das Signal S gegen die Konzentration c aufgetragen |
| Wiederfindungsrate | recovery | Ausbeute der Probevorbereitung Überprüfung, ob bei der Probeaufarbeitung (z. B. Derivatisierung, Extraktion, Injektion etc.) ein Teil der Substanz “verschwindet“ |
| Selektivität | selectivity | Störung durch Begleitstoffe Fähigkeit eines Analyseverfahrens verschiedene Komponenten nebeneinander zu bestimmen |
| Robustheit | robustness | Störanfälligkeit durch veränderte Bedingungen (Analysenparameter, Gerät, Labor usw.) |
| Nachweisgrenze | limit of detection | Kleinste nachweisbare Menge „Ja/Nein“-Entscheidung |
| Bestimmungsgrenze | limit of quantitation | kleinste quantifizierbare Menge „Wieviel“-Entscheidung: |
Kurzdefinitionen Prüfparameter
| Bezeichnung | Aussage über |
|---|---|
| Richtigkeit | systematische Fehler |
| Präzision • Wiederholbarkeit (Wiederholpräzision) • Vergleichbarkeit (Vergleichspräzision) | zufällige Fehler laborintern verschiedene Labors |
| Robustheit • Verfahrensstabilität („robustness“) • Übertragbarkeit („regedness“) (Wiederholbarkeit, Vergleichbarkeit) | Abhängigkeit von variierenden Bedingungen Störanfälligkeit durch veränderte |
| Selektivität | Fähigkeit zur Bestimmung mehrerer Komponenten nebeneinander |
| Wiederfindungsrate | Ausbeute der Probenvorbereitung |
| Linearität | Abhängigkeit Signal/Konzentration |
| Linearität | linearity |
| Nachweisgrenze | kleinste nachweisbare Menge (Konzentration) |
| Bestimmungsgrenze | kleinste quantifizierbare Menge (Konzentration) |
| Spezifität | Störanfälligkeit gegenüber Begleitkomponenten |
| Messbereich, („range“) dynamischer Arbeitsbereich | Konzentrationsbereich für erlaubte quantitative Aussagen |
| Unsicherheit, Vertrauensintervall | Schwankungsbereich des Analysenergebnisses (Messwertes) |
| Reproduzierbarkeit | Wiederholpräzision innerhalb kurzer Zeitabstände |
| Genauigkeit | Oberbegriff für Richtigkeit und Präzision |
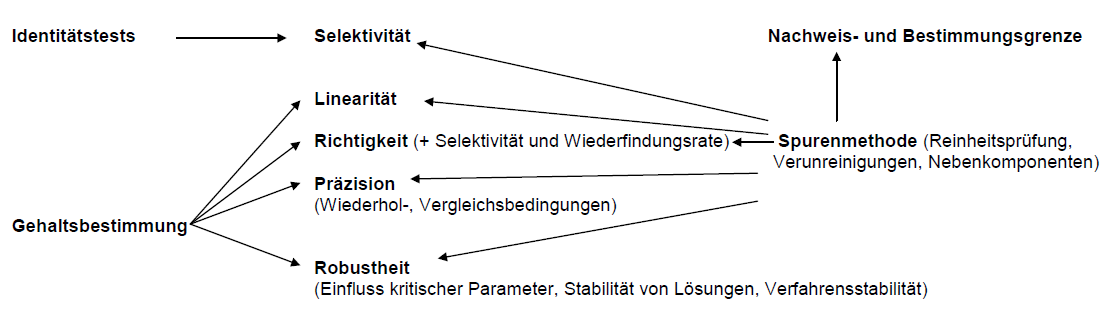
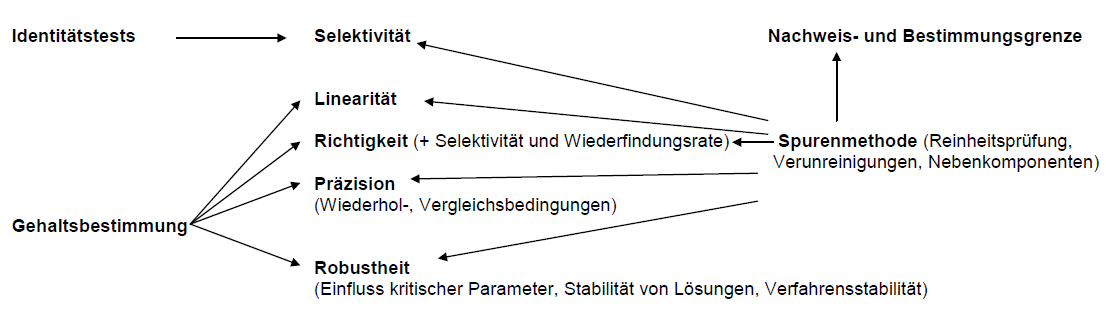
Umfang der Methodenvalidierung in der Analytik
Fließschema zur Methodenvalidierung einer „Spurenmethode“ mittels Chromatographie
(Reinheitsprüfung, Neben- und Abbauprodukte)
I am text block. Click edit button to change this text. Lorem ipsum dolor sit amet, consectetur adipiscing elit. Ut elit tellus, luctus nec ullamcorper mattis, pulvinar dapibus leo.
Fließschema zur Methodenvalidierung von Identitätstests in der Chromatographie
Qualitätsregelkarten
Kontrollkarten als Qualitätssicherungselement
Jedes Routinelabor muss seine Befähigung und Eignung nachweisen, indem es aufzeigt, dass es zuverlässig „richtige“ Werte mit einer bekannten und akzeptablen Präzision bestimmt. Dieser Nachweis kann sowohl durch die Teilnahme an Ring bzw. Vergleichsversuchen (externe Qualitätssicherung) als auch durch Führen von Kontrollkarten (interne Qualitätssicherung) erbracht werden.
Qualitätssicherung im Labor
Zur Überwachung der Zuverlässigkeit von Prozessen, insbesondere im Produktionsbereich, werden diese so genannten Regelkarten schon seit langem verwendet. Mit der zunehmenden Bedeutung der Qualitätssicherung im Labor werden sie immer stärker auch in der Analytik eingesetzt. Der zu überwachende Prozess ist hier das analytische Messverfahren selbst.
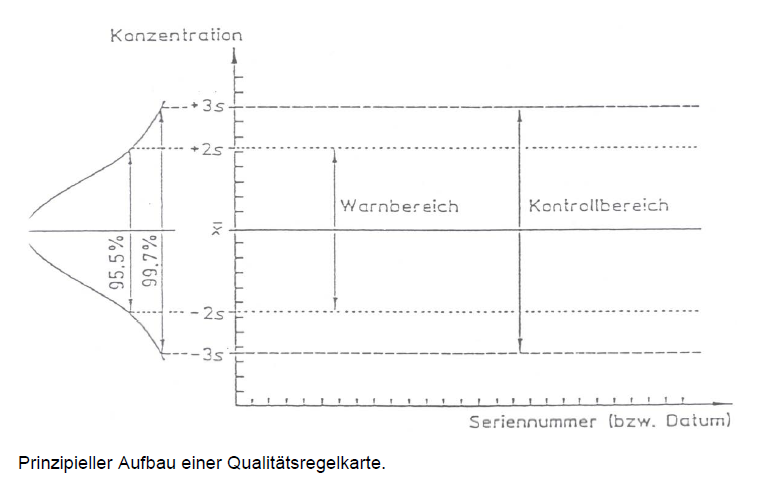
Die Abbildung zeigt beispielhaft den Aufbau einer Qualitätsregelkarte.
Prinzipieller Aufbau einer Qualitätsregelkarte (oder einfach: Kontrollkarte)
Was kann in eine Kontrollkarte eingetragen werden
In dieser Karte können die Messergebnisse einer stets gleichen Kontrollprobe, die zusammen mit den Routineproben analysiert wird, eingetragen werden. Messwerte oder daraus ermittelte Kennwerte, wie z. B. Streubreite aus Mehrfachbestimmungen, werden gegen die Zeit aufgetragen. So wird anhand weniger „Spielregeln“ die visuelle Beurteilung des Prozesses und gegebenenfalls die Einleitung von Überprüfungs- oder Korrekturmaßnahmen ermöglicht.
Nachweis der Funktion eines Prozesses
Gleichzeitig kann das Funktionieren eines Prozesses nachgewiesen und damit das System im aktuellen Zustand validiert werden.
Die entscheidenden Vorteile der Kontrollkartentechnik sind:
Vorteile einer Kontrollkarte
• Alle Mitarbeiter treffen ohne subjektive Einflüsse dieselben Entscheidungen
• Korrigierende Einflüsse können schnellstmöglich veranlasst werden
• Retrospektive Beurteilungen des Systemzustandes sind leicht möglich (Qualitätsnachweis)
• Das Auftreten von systematischen Fehlern und Trends kann visuell leicht und zeitnah erkannt werden
Voraussetzungen für das Führen einer Kontrollkarte
Selbstverständlich ist für den Einsatz von Kontrollkarten in der Analytik Voraussetzung, dass die Analysenprozesse sich sporadisch oder regelmäßig wiederholen, d. h. dass derselbe Analyt in einer möglichst wenig veränderten Matrix in einem überschaubaren Konzentrationsbereich immer wieder bestimmt wird.
Einsatzgebiete wichtigste Arten von Kontrollkarten
Einsatzgebiete sind daher vor allem die Freigabe von Chargen eines Produktionsprozesses, die routinemäßige Analytik in medizinischen Laboratorien und die regelmäßige Kontrolle bestimmter Abwässer. Es gibt verschiedene Arten von Kontrollkarten. Im analytischen Labor sind neben der mit Abstand am wichtigsten Mittelwertkontrollkarte außerdem die Wiederfindungs-, die Blindwert- sowie die Spannweitenkontrollkarte von Bedeutung. (Im Folgenden sei mit dem Begriff Kontrollkarte stets die Mittelwertkontrollkarte gemeint, sowie nicht anderes erwähnt.)
Präzisionskontrolle
Grundsätzlich können Kontrollproben sowohl zur Erkennung zufälliger Fehler (Präzisionskontrolle) als auch systematischer Fehler (Richtigkeitskontrolle) verwendet werden. Bei der Präzisionskontrolle werden die Werte der Kontrollprobe mit den zuvor gemessenen Werten der Kontrollprobe mit den zuvor gemessenen Werten derselben Probe verglichen.
Richtigkeitskontrolle
Bei der Richtigkeitskontrolle werden die Werte mit einem gegebenen Bezugswert verglichen. Der zu überprüfende Messwert (meist der Gehalt an Analyt) muss bei einer Richtigkeitskontrolle also bekannt sein. Wegen des dazu erforderlichen Messaufwandes bei der Herstellung sind Richtigkeitskontrollproben grundsätzlich teurer als Präzisionskontrollproben.
Vorperiode
Während einer Vorperiode werden zunächst Daten gesammelt, aus denen über Mittelwerte und Standardabweichung die Warn- und Eingriffsgrenzen ermittelt werden. Diese Daten können auch zur Validierung der Methode verwendet werden. Üblicherweise umfasst eine Vorperiode zwanzig Werte. Erst danach kann eine Qualitätsregelkarte genutzt werden. Wichtig ist, dass die Prüfbedingungen in der Vor- und der anschließenden Kontrollperiode vergleichbar sind.
Kontrollperiode Warngrenze Eingriffsgrenze
Als Warngrenze wird zumeist ein Band der Breite 4s (Mittelwert ± 2s, mit s als Standardabweichung der Vorperiode), als Eingriffsgrenze ein Band der Breite 6s (Mittelwert ± 3s) festgelegt.
Annahmekarten
Im Idealfall, d. h. der Prozess befindet sich unter statistischer Kontrolle, und lediglich zufällige, aber keine systematischen Fehler sind wirksam, befinden sich im ersten Band 95,5 % und im letzteren 99,7 % aller Messwerte. Sind bereits Forderungen, z. B. aus Spezifikationen, vorhanden, so werden durch diese die Eingriffsgrenzen bzw. Grenzwerte festgelegt während die Warngrenzen entfallen. Man spricht in diesem Fall von Annahmekarten bzw. Annahme-Qualitätsregelkarten.
Außer-Kontroll-Situation
Beim Auftreten von Außer-Kontroll-Situationen müssen besondere Maßnahmen ergriffen werden. Dies kann im einfachsten Fall eine visuelle Systemüberprüfung oder eine Plausibilitätsprüfung der Ergebnisse sein, in schweren Fällen aber auch zum Sperren bzw. der Reparatur eines Gerätes führen. Die Außer-Kontroll-Situationen stellen also die Spielregeln dar, die besondere Maßnahmen unabhängig vom persönlichen Ermessen des Operators auslösen. Als Außer-Kontroll-Situationen gelten:
• ein Wert außerhalb der Kontrollgrenzen
• sieben Werte in Folge ansteigend bzw. abfallend
• sieben Werte in Folge über bzw. unter dem Mittelwert
• zwei von drei Werten in Folge außerhalb der Warngrenzen
Treten solche Situationen auf, so sollte – selbst wenn keine Ursache gefunden werden kann – eine schriftliche Bewertung der Qualitätsdokumentation erfolgen. Es ist nämlich durchaus nicht zwingend, dass Außer-Kontroll-Situationen auf Unregelmäßigkeiten oder Fehler hinweisen.
Average Run Length
Rein statistisch bedingt führen zufällige Fehler von Zeit zu Zeit ebenfalls zu Außer-Kontroll-Situationen, obwohl alles in bester Ordnung ist. Quantifiziert wird dieses „Risiko“ durch die Average Run Length (ARL), also die Laufzeit bis zum Auftreten einer Außer-Kontroll-Situation, wenn der Prozess sich vollkommen unter statistischer Kontrolle befindet.
Periodische Schwankungen
Gerade bei der retrospektiven Überprüfung von Vermutungen („Sind die Messwerte seit dem Wechsel der Kalibriersubstanz erhöht?“) zeigen Kontrollkarten ihren praktischen Nutzen. Zweifellos bedeutet das Führen von Kontrollkarten einen zusätzlichen Aufwand, der finanzierbar sein muss. Die zweitaufwenige Erstellung per Hand wird aber mehr und mehr durch Computerprogramme abgelöst. Von einem modernen LIMS – im Zusammenhang mit Smart Data Analysis – wird man also in Zukunft umfassende Möglichkeiten zum Führen von Kontrollkarten erwarten, wie die online-Datenübernahme von Messgeräten und eine automatische Steuerung von Maßnahmen bei Außer-Kontroll-Situationen, beispielsweise Alarmauslösung, Wiederholung einer Analyse oder Sperren eines Gerätes. Neben den Kontrollkarten ist das Schätzen der Messunsicherheit ein bewährtes, billiges, schnelles und bereits akzeptiertes Werkzeug der Qualitätssicherung.
Eine Methode wird in einzelne Schritte zerlegt, z. B. Probevorbereitung, Messung, Befundung. Der Fachmann schätzt aus den gemachten Erfahrungen usw. den Fehler der einzelnen Schritte. Hier wird zwischen zwei Extremen, dem günstigsten und dem ungünstigsten Fall, unterschieden („best/worst case“). Die geschätzten Fehler der einzelnen Schritte werden zum Quadrat genommen, die Quadrate addiert und aus der Summe die Wurzel gezogen. Der geschätzte Fehler der Methode liegt zwischen den zwei extremen Fällen („best case“, „worst case“). Diese in Kürze beschriebene Möglichkeit eignet sich für einmalige Fragestellungen, z. B. im F+E-Bereich. Genaueres zum Schätzen der Messunsicherheit findet sich in: Kromidas, Validierung in der Analytik, Wiley-VCH.
Retrospektive Überprüfungen
Eine Kontrollkarte verhält sich sozusagen wie eine Alarmanlage, bei der eine hohe Ansprechempfindlichkeit stets durch die ebenfalls erhöhte Gefahr von Fehlalarmen erkauft wird. Der Wert einer Kontrollkarte erweist sich aber nicht erst im Auftreten von Außer-Kontroll-Situationen. Nützliche Informationen z. B. über periodische Schwankungen oder eine Verringerung der Streubreite lassen sich aus ihnen ablesen.
Trends auf dem Gebiet der analytischen Validierung
Fazit:
• Verstärkter Fokus auf praxisnahe Prüfung, ob also besagte Methode für die Routine tatsächlich geeignet ist. Das bedeutet beispielsweise, dass Risikobewertung und Robustheit immer wichtiger werden. Auch eine kontinuierliche Überprüfung (Monitoring) während des gesamten Lebenszyklus der Methode wird immer mehr verlangt
• Gegenläufige Tendenzen: Während seitens bestimmter Organisationen einerseits ein pragmatischer Blick (siehe erster Punkt) forciert wird, wird andererseits gleichzeitig von bestimmten nationalen Behörden eine sture, oft unsinnige Validierungs-Praxis verlangt
Kommentare, Hinweise
Zwei Zitate/Textpassagen bzw. häufig verwendete Begriffe, welche den stärker werdenden pragmatischen Ansatz untermauern:
1. „continual assurance that the process (analytical procedure) remains in a state of control (the validate state) during commercial manufacture“
2.
• Analytical Procedure Lifecycle Management (APLM)
• Procedure Performance Qualification (PPQ)
• Process stability and capability
• Requirements for routine process monitoring of analytical procedures
• Trending as part of the analytical control strategy and confirmation of the ATP
• „matrixbased“ calibration
• „matrixbased“ quality control
Bis dato: Methode gut? Ergebnis gut
Zukünftig: Anforderungen klar definiert? Eine Methode, die den Anforderungen genügt, ist gut (geeignet)
Merke: Der Formulierung der „Anforderungen“ fällt eine gewichtige Bedeutung zu! So gehört u. a. folgendes dazu:
• Risikobewertung (z. B. Änderungen-Auswirkungen)
• Methode bedeutet: Vollständiges (!) Verfahren
• fundiertes Wissen über Robustheit
• Geeignete Systemeignungstests
• Analytischer Transfer ist der selbstverständliche Schritt nach einer Validierung: Eine Selbstverständlichkeit
• Monitoring („Continued Method Performance Verification“)
Zur Praxis der Validierung heute
Bis dato: Methode gut? Ergebnis gut
Zukünftig: Anforderungen klar definiert? Eine Methode, die den Anforderungen genügt, ist gut (geeignet)
Merke: Der Formulierung der „Anforderungen“ fällt eine gewichtige Bedeutung zu! So gehört u. a. folgendes dazu:
- Risikobewertung (z. B. Änderungen-Auswirkungen)
- Methode bedeutet: Vollständiges (!) Verfahren
- fundiertes Wissen über Robustheit
- Geeignete Systemeignungstests
- Analytischer Transfer ist der selbstverständliche Schritt nach einer Validierung: Eine Selbstverständlichkeit
- Monitoring („Continued Method Performance Verification“)
Über eine zeitgemäße Validierung – das „4-Phasen-Modell“
- Was soll die Methode können?
- Auswahl der Methode, evtl. Methodenentwicklung und Optimierung
Merke:- Methode bedeutet „Prüfverfahren“
- Risikobewertung, Robustheit etc. d.h. Charakteristika
des Verfahrens kennen lernen, klar identifizieren (optimal)
- Methoden-Qualifizierung (ehemals „Validierung“)
Merke:- Überprüfung unter Routinebedingungen
- analytischer Transfer – welcher Art auch immer – gehört in diese Phase
- Kontinuierliche Methoden-Verifizierung
- Monitoring; Absicherung, dass Methode weiterhin „OK“ ist
z. B. SST-Daten, SPC mit Präzisionsdaten aus
Stabilitätsstudien, haben etwaige Änderungen einen (signifikanten!)
Einfluss auf das Ergebnis?
- Monitoring; Absicherung, dass Methode weiterhin „OK“ ist
Literaturliste zum Thema (Auswahl)
(Sehr guter Überblick über DIN: Beuth-Verlag)
- Lothar Sachs, Jürgen Hedderich: „Angewandte Statistik“, Springer Verlag, ISBN 3540321608
- Herbert Feltkamp, Peter Fuchs, Heinz Sucker: „Pharmazeutische Qualitätskontrolle“, Thieme Georg Verlag, ISBN 3136115015
- Herbert Feltkamp, Peter Fuchs, Heinz Sucker: „Pharmazeutische Qualitätskontrolle“, Thieme Georg Verlag, ISBN 3136115015
- Stavros Kromidas: „Validierung in der Analytik“, Wiley-VCH Verlag, 2. überarbeitete Auflage, ISBN 3527329390
- Stavros Kromidas: „Validierung in der Analytik“, Wiley-VCH Verlag, 2. überarbeitete Auflage, ISBN 3527329390
- Thomas Schreppe, Rainer H. Müller: „Qualitätsmanagement und Validierung in der pharmazeutischen Praxis“, Editio Cantor Verlag, ISBN 3871932698
- Joachim Ermer, John Miller (Eds.): „Method Validation in Pharmaceutical Analysis“, Wiley-VCH, ISBN 3527312552
- Michael Hiob, Qualifizierung und Validierung aus Behördensicht: GMP-konforme Umsetzung des Annex 15 (GMP-Fachwissen), GMP Verlag, ISBN 3958070744
- Anforderungen der US-FDA an die Computervalidierung, GMP Verlag, ISBN 978-3-95807-082-0
- Stephen Robert Goldman: „Handbook of Computer and Computerized System Validation for the Pharmaceutical Industry“, Kindle Edition, ASIN B0017GTRTG
- Gil Bismuth, Shosh Neumann: „Cleaning Validation: A Practical Approach“, Informa HealthCare, ISBN 1574911082
Regelwerke in der AQS und Statistik (Auswahl)
| DIN 32645 | Nachweis-, Erfassungs- und Bestimmungsgrenzen |
| DIN 38402 A41 | Ringversuche |
| DIN 38402 A42 | Ringversuche und statistische Auswertung |
| DIN 38402 A51 | Kalibrierung von Analysenverfahren |
| DIN 38402 A51 | Gleichwertigkeit zweier Analysenverfahren |
| DIN 55350 | Begriffe des QS und Statistik |
| ISO 5725 | Accuracy (trueness and precision) |
| ISO 8402 (wurde durch ISO 9000 ersetzt) | Qualität , Begriffe |
| ISO 9000 | Qualitätsmanagement |
| ISO 9001 | QS-System – Modell zur Darlegung von QS in Entwicklung, Produktion und Kundendienst |
| ISO 9002 | QS-System-Modell in der Montage und Produktion |
| ISO 9003 | QS-System – Modell in der Endprüfung |
| ISO 9004 | Qualitätsmanagement und Elemente der QS – ein Leitfaden |
| DIN ISO 17025:2018 | Richtlinien zum Betreiben von Prüf- und Kalibrierlaboratorien |

