Startseite » Infos für Fortgeschrittene » 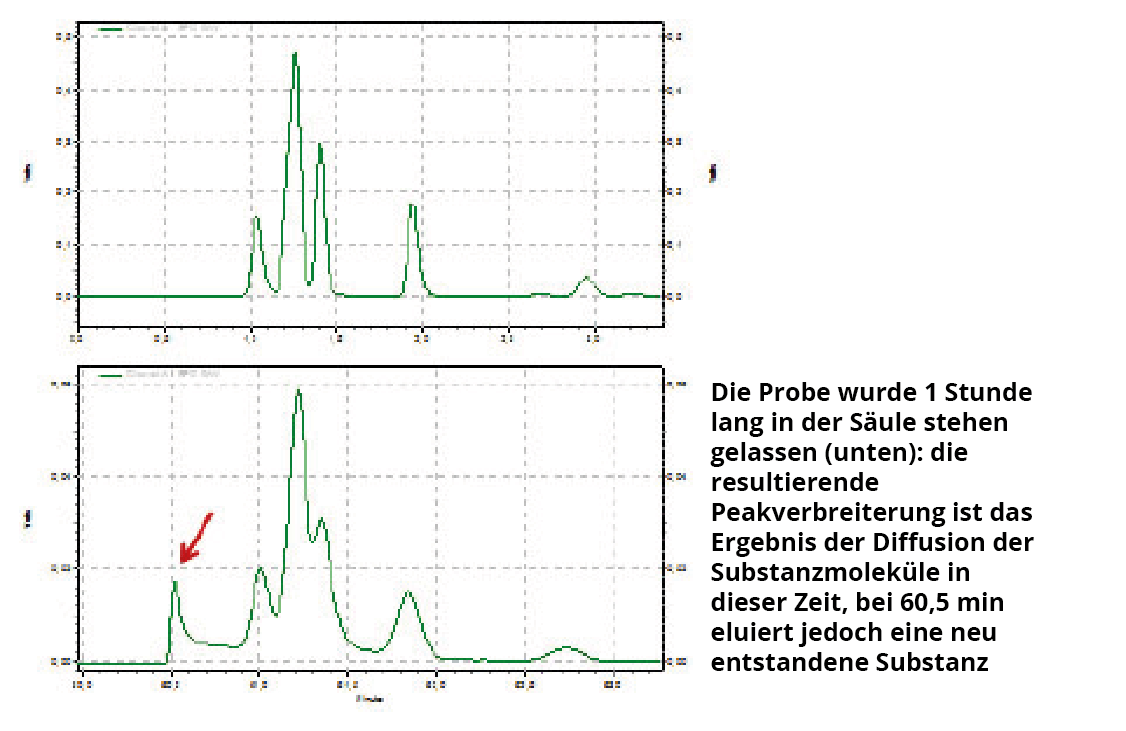


Einführung in die Thematik und Annahmen
Nachfolgend beschriebenes Konzept basiert auf den Erfahrungen aus unterschiedlichen Projekten zur Methodenentwicklung und Optimierung. Dabei geht es weniger darum, diese Vorschläge „1 zu 1“ zu übernehmen, vielmehr sollen diese nur Anregungen sein; die sich tatsächlich ergebende Vorgehensweise kann nur das Resultat von realen Gegebenheiten vor Ort sein. Die Variationen geben einerseits die individuellen Präferenzen in den beteiligten Laboren und andererseits Unterschiede in der vorhandenen Hardware wieder. Am Ende des Beitrages findet sich ein Résumé.
Vorbemerkungen
- Es wird unterstellt, dass eine moderne Gradientenanlage mit 2-4 Lösungsmitteleingängen bzw. entsprechendem Eluentenschaltventil, (kühlbarem) Säulenofen, Säulenschaltventil und PDA (Photo Dioden Array–Detektor) zur Verfügung steht; eine LC-DAD-MS-Kopplung wäre zweifelsohne von Vorteil. Obschon ein 12-Wege Säulenschaltventil naturgemäß mehr Variationsmöglichkeiten bietet, wird im nachfolgenden Vorschlag das 6-Wege Säulenschaltventil berücksichtigt, da jenes die größte Beliebtheit bei den Anwendern genießt.
- Es wird ferner aus Gründen der Ökonomie – zumindest für die ersten Orientierungsversuche, siehe weiter unten – dringend empfohlen, mit kurzen Säulen (20-50 mm, 2-3 µm) zu beginnen. So könnten auch an klassischen HPLC-Geräten mit Hilfe von schnellen Läufen in kürzester Zeit Trends sowie geeignete chromatographische Parameter wie pH-Wert, Lösungsmittel und Gradient grob ermittelt werden. Alternativ sollte natürlich, falls die Möglichkeit besteht, eine UHPLC verwendet werden. Erwartet man mehr als 20-30 Peaks und/oder liegt eine „schwierige“ Matrix vor, wäre in etwa folgende Säulendimensionierung empfehlenswert: 125-150 x 3-4 mm, 5 µm.
- In dem weiter unten vorgestellten Schema wird von „Worst Case“ ausgegangen, dass nämlich keinerlei Informationen über die Probe vorliegen. Im Falle von bekannten oder erwarteten Substanzen in der Probe, sollten mit einem vertretbaren Aufwand Informationen über deren chromatographisches Verhalten eingeholt oder ggf. Substanzdaten wie pKs-/log P-Wert ermittelt werden. Daraus könnte u. U. eine erste Entscheidung bzgl. Säulentyps und Eluenten getroffen werden, z. B. ein kleiner pKs-Wert macht den Einsatz von säurestabilen Säulen notwendig. Schließlich wird vorausgesetzt, dass für den Einsatz von kurzen Säulen keine Bedenken bestehen. Ansonsten erhöht sich der unten angegebene Zeitbedarf entsprechend.
Systematische Vorgehensweise
1. Schritt: Orientierungsversuche (Tagesexperiment)
Man beginnt mit drei schnellen, linearen Gradienten (z. B. 20-30% bis 95% Acetonitril) bei drei pH-Werten (z. B. 3, 4,5, 7) an sechs unterschiedlichen Säulen, z. B. zwei hydrophobe (etwa C18 Hersteller 1, C18 Hersteller 2), zwei polare (etwa Biphenyl/Phenyl-Hexyl und/oder EPG, EPG: Phase mit eingebauten polaren Gruppe), zwei stark polare Säulen wie Mixed Mode und PFP (zur Auswahl von HPLC-RP-Säulen, siehe „Colona“). Bei einer angenommenen (und zunächst ausreichenden) Gradientendauer von ca. 10 min ergibt sich in etwa folgender Zeitbedarf: 3 pH-Werte x 6 Säulen x ca. 14 min (10 min Trennzeit plus 4 min Equilibrierzeit) = ca. 250 min, also ca. 4-5 Stunden. An der „besten“ Kombination Säule-pH-Wert (Kriterium: An erster Stelle Anzahl und an zweiter Stelle Form der Peaks) wird nun wie folgt variiert:
- Der pH-Wert wird mit Hilfe einer zweiten Säure/Base eingestellt (z. B. statt Phosphorsäure, nun Ameisen- oder Perchlorsäure, statt Natronlauge nun Ammoniak oder Triethylamin. Dieser Schritt zur Feinoptimierung kann auch später erfolgen.
- 50% des Acetonitril-Anteils im Eluenten werden gegen 50% Methanol (z. B. statt eines Gradienten von 30% auf 95% Acetonitril, nun von 15% ACN plus 15% MeOH auf 47,5% ACN plus 47,5% MeOH) bzw. gegen 10% THF ausgetauscht. Zeitbedarf für diese drei Läufe: ca. 30 min.
Nach diesem ersten Schritt, der ca. 5 – 5,5 Stunden Messzeit in Anspruch nimmt (realistisch: Ein Tag), verfügt man über folgende Informationen: An welchem Phasentyp erhält man bei welchem pH-Wert mit welchem Modifier und mit welchem organischen Lösungsmittel im Eluenten die größte Anzahl an Peaks? Sollte sich andeuten, dass die beste Selektivität im Alkalischen zu erwarten ist, sollten bei drei höheren pH-Werten (z. B. 10, 11, 12) und mit Hilfe von zwei Modifiern (z. B. Ammoniak, Ammoniumcarbonat oder Boratpuffer) sechs alkalistabile Säulen getestet werden, z. B. Gemini/NX, YMC Triart, Zorbax Extend, XBridge C18/Shield, Kromasil EternityXT, Hypercarb. Hinweis: Bei bestimmten Applikationen (Ionenaustausch, RIDetektion, Gelfiltration) werden mithilfe eines Eluentenschaltventils/Niederdruckventils natürlich isokratische Lösungsmittelgemische mit Variation je nachdem des Lösungsmittels, des pH-Wertes oder des Salzgehaltes über die sechs Säulen geschickt. Selbstverständlich gibt es mehrere Varianten für diesen ersten Schritt, nachfolgend beispielhaft eine:
Variante
Schritt 1
Mit Hilfe von schnellen Gradienten, siehe weiter oben, testet man drei Säulen (z. B. C18, EPG, Mixed Mode/PFP) bei je einem Lösungsmittel (üblicherweise Acetonitril und Methanol bzw. statt reinem Methanol, sinnvollerweise 50% Acetonitril/50% Methanol) und je drei pH-Werten, ansonsten gleiche Vorgehensweise wie weiter oben beschrieben. Anzahl der Läufe: 3 Säulen x 2 Lösungsmittel x 3 pH-Werte = 18 Läufe, Zeitbedarf: 18 Läufe x 14 min (10 min Trennzeit plus 4 min Equilibrierzeit) = ca. 4 Stunden.
Schritt 2
Nach diesen Läufen wird anhand der Chromatogramme entschieden, welcher Parameter die größte Variation zeigt, das Lösungsmittel oder der pH-Wert? Anschließend werden entweder bei zwei pH-Werten und dem „besseren“ Lösungsmittel oder bei dem „besten“ pH-Wert und zwei Lösungsmittelzusammensetzungen drei weitere Säulen getestet. Anzahl der Läufe: 3 Säulen x 2 pH-Werte oder 3 Säulen x 2 Lösungsmittelzusammensetzungen = 6 Läufe. Zeitbedarf: 6 Läufe x 14 min (10 min Trennzeit plus 4 min Equilibrierzeit) ca. 1,5 Stunden.
Schritt 3
Schließlich wird die „beste“ Säule beim „optimalen“ pH-Wert und dem „besseren“ Lösungsmittel einem letzten kleinen Optimierungsschritt unterzogen: Man kombiniere je zwei Gradienten (z. B. lang + niedriger Fluss, und steil + hoher Fluss, Infos zur Gradientenoptimierung, siehe Buchbeitrag aus HPLC-Tipps Band 3) zwei Temperaturen (z. B. 15°C und 35°C), das ergibt vier Läufe. Zeitbedarf: 4 Läufe x 14 min, siehe weiter oben, ca. eine Stunde. Gesamt-Zeitbedarf für die drei Schritte: 4 + 1,5 + 1 = 6,5 Stunden. Berücksichtigt man auch einen Zeitpuffer für die Temperatureinstellung sowie allerlei kleinere Tätigkeiten, wären diese Tests wahrscheinlich an einem Tag durchführbar. Auch hier werden Trends erkannt und man erhält ähnliche Informationen wie oben: An welchem Phasentyp erhält man bei welchem pH-Wert mit welchem organischen Lösungsmittel bei welchem Gradienten und bei welcher Temperatur die größte Anzahl an Peaks?
2. Schritt: Säulenauswahl (Nachtexperiment)
Das Säulenschaltventil wird mit der „besten“ der sechs getesteten Säulen als Referenzsäule und fünf weiteren, ähnlichen Charakters, bestückt. Die sechs Säulen werden über Nacht mit zwei Gradienten (siehe weiter oben) bei einem pH-Wert von +/- 0,5 vom bereits als „optimal“ festgestellten pH-Wert gefahren: 2 pH-Werte x 6 Säulen = 12 Läufe.
3. Schritt: Feinoptimierung, Methodenrobustheit (Tagesexperiment)
Die bis dato „beste“ Kombination Eluent, pH-Wert und Säule wird nun einer Feinoptimierung/Justierung unterzogen: Variation von Gradient (Anfangs-, Endbedingungen, Steigung, Gradientenvolumen und -profil), Temperatur und ggf. Teilchengröße und Säulenlänge. Zu diesem Zeitpunkt sollte aus ökonomischen Gründen – da ja chromatographische Parameter sowieso systematisch variiert werden – eine erste Überprüfung der Methodenrobustheit erfolgen (Einfluss von z. B.
kleinen pH-Wert-Änderungen usw.). Es sollte der Vollständigkeit halber erwähnt werden, dass für derartige Optimierungsschritte wie auch Experimente zur Methodenrobustheit kommerzielle Optimierungsprogramme (DryLab, ChromSword usw.) bewährte, leistungsfähige Tools darstellen. Aber auch ohne solche Hilfen ist durch eine systematische Vorgehensweise realistisch, dass nach ca. zwei Tagen und einer Nacht die Methode grob „steht“. Spätestens an dieser Stelle sollten die Ergebnisse mit einer(m) nicht in den Versuchen involvierten Fachfrau/Fachmann kritisch diskutiert werden. Die ersten drei Hauptschritte dieses Procedere sind beendet, man ist mit den Ergebnissen evtl. grob zufrieden, die Feinoptimierung kann sich nun anschließen.
Empfehlung:
Besteht ein begründetes Interesse an „Wahrheit“, so sollte Schritt vier folgen. Soll die betroffene Methode später in der Routine möglichst zuverlässig funktionieren, so sollte Schritt fünf folgen.
4. Schritt: Überprüfung der Peakhomogenität, „cross-Experimente“ (Nachtexperiment)
Die Peakhomogenität kann grundsätzlich mit Hilfe der Spektroskopie und von orthogonalen Techniken überprüft werden. Neben des PDA eignet sich eher die MS- (ESI-MS, Q-TOF, MALDI) und die NMR Off-Line/On-Line-Kopplung. 2D-Trennungen – häufig gekoppelt mit spektroskopischen Techniken – haben sich in letzter Zeit zu dem Tool „par excelence“ entwickelt, wenn es um die „ultimative“ Überprüfung der Peakhomogenität bzw. um die Trennung von sehr komplexen Proben und/oder komplexen Matrices geht („Trends in der HPLC“). Welcher Aufwand gerechtfertigt ist, kann nur individuell entschieden werden. Es scheint so zu sein, dass orthogonale Experimente („cross-Experimente“) auch ohne aufwendige Kopplungstechniken eine ausreichende Sicherheit bieten. Eine Zuordnung der Peaks ist zunächst nicht notwendig, es geht lediglich um deren Anzahl. Nachfolgendes Schema gibt einen Überblick über einfache bis hin zu aufwendigeren Möglichkeiten zur Überprüfung der Peakhomogenität wieder. Bemerkung: Es wird unterstellt, dass für das aktuelle Trennproblem die optimale Hardware (Kapillaren, Detektorzelle) und die optimalen Einstellungen gewählt wurden, z. B. Referenzwellenlänge, Datenrateaufnahme, Bandbreite, Slit, Zeitkonstante bzw. bei LC-MS dwell-time usw.
Überprüfung der Peakhomogenität in der RP-HPLC
- Zunächst bei konstanter chromatographischen Auflösung apparative Möglichkeiten ausnutzen ohne die Methodenparameter zu verändern: PDA, ggf. LC-MS (optimal: Hochauflösende LC-MS und unterschiedliche Ionisierungstechniken) .
- Bei isokratischen Läufen kann folgendes überprüft werden: Befinden sich die Quotienten Peakbreite/Retentionszeit aller Peaks auf einer Geraden? Im Falle eines Ausreißers besteht Verdacht auf Inhomogenität des entsprechenden Peaks.
- Einfache und schnelle Überprüfungen: Injektionsvolumen verringern, Probe mit Wasser/Eluent verdünnen/mit Neutralsalz versetzen und erneut injizieren, gleiche stationäre Phase mit kleineren Teilchen einsetzen.
- Orthogonale Tests (das beste Tool bzgl. Aufwand/Nutzen):
- Gleiche Säule, anderer Eluent (z. B. statt Acetonitril, nun Methanol bzw. statt pH-Wert X, nun pH-Wert Y)
- Gleicher Eluent, andere Säule (z. B. statt einer polaren, nun eine apolare stationäre Phase verwenden)
Zum Beispiel: Probe verdünnen, eine ganz andere Säule/Eluent wählen und bei 15°C über die Mittagspause laufen lassen (evtl. Gradientenvolumen erhöhen, Buchbeitrag aus HPLC-Tipps Band 3)
- Hauptpeak fraktionieren (vordere Flanke, Peakspitze, hintere Flanke), das Einengen der Fraktionen dürfte selten notwendig sein.
a) Die Fraktionen werden erneut einzeln injiziert
b) Die Fraktionen werden erneut einzeln injiziert, nachdem eine zweite Säule in Serie geschaltet wurde
c) Die Fraktionen werden mit Hilfe der GC, DC/AMD oder CE untersucht
d) Die Fraktionen werden mit Hilfe der IR-, MS,- NMR-Spektroskopie untersucht
Welcher Aufwand lohnt sich?
Die Tests 1 bis einschließlich 4 bzw. 5a oder 5b sollten bei wichtigen Proben stets durchgeführt werden, möchte man sich der „Wahrheit“ annähern. Der Aufwand von ca. einem Tag – für Prüfpunkt 1 bis 4 – bzw. von ca. zwei Tagen für zusätzlich Prüfpunkt 5a und b erscheint angemessen; ob der Aufwand für Prüfpunkt 5c-d auch noch gerechtfertigt ist, kann nur individuell entschieden werden.
5. Schritt: Methodenrobustheit (Tagesexperiment) und Säulenstabilität („Wochenendexperiment“)
Ein letzter Schritt eines Methoden-Entwicklungsprojektes sollte die Überprüfung der Robustheit sein – falls die betroffene Methode nicht für eine einmalige Anwendung gedacht ist. Der Aufwand und der Umfang hängen naturgemäß von der konkreten Fragestellung ab. „Robustheit“, obschon sehr wichtig, stellt kein Thema des vorliegenden Beitrages dar, deswegen werden nachfolgend lediglich stichwortartig einige mögliche Tests genannt (Näheres, siehe „Überprüfung der Robustheit“).
Dringender Hinweis:
Die Überprüfung der Robustheit sollte unbedingt mit realen Proben erfolgen. Sollten solche nicht vorliegen, wären Proben anzusetzen, die den später in der Routine Eingesetzten möglichst ähnlich sind:
Matrix/Placebo/Lösungsmittel/Hilfsstoffe/Stresslösungen plus Analyt. Es macht wenig Sinn, die Robustheit von Methoden mit Standardlösungen überprüfen zu wollen.
A. „Verträglichkeit“ Probe-stationäre Phase
- Katalytische Wirkung des Kieselgels? Kieselgel, als stationäre Phase oder als Matrix bei RP-Phasen, ist ein sehr guter Feststoffkatalysator. Es zeigt sich immer wieder, dass viele kleine Peaks im Chromatogramm Substanzen darstellen, die „in situ“ in der Säule durch die katalytische Wirkung des Kieselgels entstanden sind und nicht ursprünglich in der Probe vorhanden waren. Überprüfung: Man injiziere die Probe wie gewohnt und während sich die Probe in der Säule befindet, schaltet man die Pumpe aus. Nun lässt man die Probe ca. 30-45 min lang in der Säule stehen und schaltet anschließend die Pumpe erneut an. Frage: Bleiben die Anzahl der Peaks und die Peakflächen gegenüber der „normalen“ Injektion konstant? Siehe dazu Abbildung 1: Dort wird gezeigt, dass im vorliegenden Fall in der Säule durch die katalytische Wirkung des Kieselgels eine neue Substanz entstanden ist.
Abb. 1: Überprüfung der katalytischen Wirkung des Kieselgels auf die Stabilität von Substanzen, Details, siehe Text
- Irreversible Sorption an der stationären Phase?
- Nachdem die Säule bereits equilibriert worden ist, injiziere man mehrmals die Probe. Frage: Bleibt die Peakfläche konstant?
- Man injiziere ein kleines Probenvolumen ohne Säule. Statt einer Säule wird ein Verbindungsstück zwischen Probengeber und Detektor verwendet. Ohne Säule erscheint natürlich nur ein Peak, man notiere die Peakfläche. Die Säule wird nun eingebaut und das gleiche Probenvolumen wird erneut injiziert. Man erhält je nach Selektivität 1, 2, 3… Peaks. Nun werden alle Peakflächen addiert und man vergleicht die Peakfläche(n) mit/ohne Säule. Frage: Ergibt sich eine Differenz von
kleiner als ca. 5-8%? Wenn ja, handelt sich womöglich um Messfehler bzw. es findet keine nennenswerte irreversible Sorption von Probenbestandteilen an der stationären Phase statt.
B. Einfluss des Probenlösungsmittels
Das Probenlösungsmittel bzw. das Probenmedium (Matrix, Hilfsstoffe etc.) kann die Retentionszeit, die Peakform und die Peakfläche beeinflussen. Man variiere in der Probenlösung je nachdem folgende Parameter im relevanten Bereich: pH-Wert, organischen Anteil, Matrix, Konstitution der Probe, Luftgehalt im Probenlösungsmittel. Werden Betriebsmuster analysiert, sollte man im Falle von problematischen Proben einen engen Kontakt mit den dortigen Kollegen pflegen, um stets aktuelle Informationen zu bekommen: „Kleine“ Variationen von der Probenziehung bis hin zum aktuellen Medium der Probe, die im Betrieb als nicht erwähnenswert erachtet werden, können in der Analytik große Schwierigkeiten bereiten.
C. „Verträglichkeit“ Eluent-stationäre Phase (Säulenstabilität)
Ein bewusst aggressiver Eluent wird übers Wochenende über die Säule gefördert – falls für den späteren Routineeinsatz der Methode ein großer Probendurchsatz geplant und somit die Säulenstandzeit von Bedeutung ist. Durch diese Prozedur kann eine Alterung der Säule simuliert werden und es ist fürwahr vernünftig, beispielsweise bereits am Montag zu erfahren, dass man evtl. eine selektive aber keine robuste/routinetaugliche Säule ermittelt hat, als dass diese Erkenntnis erst später im Routinebetrieb gewonnen wird… „Aggressive“ Bedingungen sind individuell zu definieren und wären je nachdem beispielsweise:
* Hoher Wasseranteil (z. B. 95-100%)
* extreme pH-Werte (z. B. pH 1,5/9,5)
* hoher Salzgehalt (z. B. 50-100 mMol)
* erhöhter Fluss (z. B. 2,5-3,5 ml/min)
* hohe Temperatur (z. B. ca. 50 C)
oder im extremen Fall alle diese Bedingungen simultan. Merke: Bei Anwendung derartiger Bedingungen über 60-70 h wird eine Alterung der Säule von ca. sechs Monaten simuliert, unter Umständen eine wichtige Information.
D. Chargenreproduzierbarkeit
Im Falle von beabsichtigten Routinemethoden sollten unbedingt 2-3 Säulenchargen („Charge“ bedeutet andere Herstellungscharge und nicht am anderen Tag gepackte Säulen…) getestet werden. Die Schritte „C“ und „D“ sind die wichtigsten, bei knapper Zeit sollte wenigstens an diese gedacht werden.
Welche Informationen gewinne ich nach einem solchen Procedere?
Zusammenfassend ergeben sich bei der bis dato beschriebenen Vorgehensweise folgende Eckdaten: Zeitbedarf: Vier Tage (realistisch: eine Woche), zwei Nächte und ein Wochenende Aufwand: Ca. 50-55 Läufe Informationen: Folgendes hätte getestet/überprüft werden können:
- 5 pH-Werte inkl. Feinjustierung
- 15 Säulen inkl. den Säulen bei den „cross-Experimenten“
- 3 organische Lösungsmittel, zwei Modifier; im Falle von starken Basen, drei weitere pH-Werte, sowie zwei Modifier und weitere Spezialsäulen
- Optimierung von Gradient und Temperatur sowie evtl. Säulendimensionierung und Teilchengröße
- Ferner hätten Informationen über die Peakhomogenität, die Methodenrobustheit und die Säulenstabilität gewonnen werden könnenSelbstverständlich gibt es zu diesem Schema eine Reihe von Varianten, es seien hier nur zwei genannt:
Variante 1
Hardwarevoraussetzung: Niederdruckgradient mit Eluentenschaltventil, d.h. sechs Lösungsmittel-Eingänge oder im Falle eines Hochdruckgradienten zwei Pumpen à drei Lösungsmitteleingänge. Statt Schritt 1-2, alternativ wie folgt:
- Nachtexperiment Ein Übersichtsgradient bei fünf pH-Werten (plus Spüllösung) und sechs Säulen (5 Gradienten x 6 Säulen = 30 Läufe)
- Tagesexperiment Die „beste“ Säule wird bei den „besten“ Bedingungen („beste“ Säure/Base, „bester“ Modifier, s.o.) nun als „Bezugs-/Referenzsäule“ mit fünf weiteren Säulen verglichen (6 Läufe).
- Nachtexperiment Sechs weitere Säulen werden bei zwei pH-Werten getestet (+/- 0,5 pH-Einheiten vom „besten“ pH-Wert): 2 pH-Werte x 6 Säulen = 12 Läufe. Schritt 3 bis 5 (Feinoptimierung und Säulenstabilität) wie oben. Zeitbedarf: Vier Tage, drei Nächte, ein Wochenende
Aufwand: Ca. 50-55 Läufe Informationen: Bei dieser Variante würden zwei pH-Werte mehr an mehr Säulen, ferner insgesamt 17 statt 11 Säulen getestet. Sie ist u. U. für „schwierige“ Proben mit vielen unterschiedlichen polaren/ionischen Spezies geeignet.
Variante 2
Hardwarevoraussetzung: Zwei quarternäre Pumpen Statt Schritt 1-2, alternativ wie folgt:
- Nachtexperiment Drei pH-Werte, drei Eluenten (je ein Lösungsmitteleingang an den zwei Pumpen ist für eine Spüllösung reserviert), sechs Säulen: 6 Eluenten x 6 Säulen = 36 Läufe
- Nachtexperiment Die bis dato „beste“ Säule wird als Bezugs-/Referenzsäule bei zwei pH-Werten (+/- 0,5 pH-Einheit vom „besten pH-Wert) und mit dem „optimalen“ Eluenten mit fünf weitere Säulen verglichen (6 Läufe): 2 pH-Werte x 6 Säulen = 12 Läufe. Schritt 3 bis 5 (Feinoptimierung und Säulenstabilität) wie oben. Zeitbedarf: Drei Tage, drei Nächte, ein Wochenende Aufwand: Ca. 50-55 Läufe Informationen: Bei dieser Variante würden die Variationen von organischen Lösungsmitteln an mehreren Säulen getestet. Sie eignet sich u. U. für Labore mit Engpässen im Personal (drei Nächte und drei Tage gegenüber zwei Nächten und vier Tagen im Falle der anfänglich beschriebenen Variante) und für Proben mit vielen, eher neutralen Komponenten.
Variationen und Alternativen
Der soeben vorgestellte Umfang für die fünf Schritte einer effektiven Methodenentwicklung/Optimierung geht vom „Worst Case“ aus. Selbstverständlich sollten die fünf Schritte je nach individueller Situation evtl. modifiziert bzw. gekürzt werden. In der Regel liegen bereits im Vorfeld diverse Informationen über die Probe vor, evtl. auch über den zukünftigen Anwendungsbereich. Jene Informationen sollten genutzt werden, um die Testbedingungen bewusst zu wählen und den Aufwand gering zu halten, z. B:
• pKS-Wert bekannt? Die Experimente mit dem pH-Wert können gezielt angegangen und deren Anzahl somit reduziert werden
• Mangelnde pH-Stabilität bei pH = X bzw. im Lösungsmittel Y? Der pH-Wert X bzw. das Lösungsmittel Y wäre zu vermeiden
• Viele Verunreinigungen in geringer Konzentration zu erwarten? Man sollte 3 µm– Material vorsehen
• Komplexe Matrix vorhanden? Der Probenvorbereitung ist größte Aufmerksamkeit zu schenken, man sollte eher 5 statt 3 µm-Material verwenden
• Sehr viele Komponenten in komplexer Matrix zu erwarten? Von Beginn an 2DChromatographie ins Auge fassen
Folgende Alternativen zum beschriebenen Konzept wären für bestimmte Fragestellungen oder in bestimmten Dienstleistungslaboren in Betracht zu ziehen:
• Vollautomatische Methodenentwicklung über Nacht (z. B. AutoChromSword)
• Superschnelle Trennungen (10-20 mm-Säule, Läufe ≤ ca. 2 min)
• Direkt die Entwicklung einer schnellen LC-MS-Methode
• Gerade ausreichende chromatographische Selektivität (Verwendung sehr kurzer Säulen) in Kombination mit spektroskopischer Spezifität (NMR, MALDI-TOF, FTIR, Röntgenfluoreszenz)
Visualisierung der Ergebnisse
Einen schnellen Überblick über erfolgreiche Kombinationen nach individuellen Kriterien könnte eine Darstellung, wie in Abbildung 2 dargelegt, verschaffen. Diverse Optimierungsparameter wie pH-Wert, Lösungsmittel, Säule usw. (siehe Tab.-Zeile 1, 2… in Abb. 2) und die entsprechenden chromatographischen Kenngrößen bzw. Resultate nach einem Optimierungsschritt (siehe Spalte 1 A-D), können in Form einer Matrix, wie in Abbildung 2 beispielhaft dargestellt, zusammengefasst werden. A bis D sind individuelle Kriterien, die nach Bedarf festgelegt werden können. Je größer z. B. der Zahlenwert der Zeile B ist, umso effektiver ist die Trennung (Peaks pro Zeiteinheit, Peakkapazität).
Abb. 2 Einfluss von Optimierungsparametern auf chromatographische Kenngrößen, Erläuterungen, siehe Text
Beispiele für individuelle Kriterien bzgl. der Güte einer RP-Trennung
A: Anzahl der Peaks absolut
B: Anzahl der Peaks pro Zeiteinheit, z. B. 4 Peaks in 10 min, 4/10 = 0,4
C: Auflösung des kritischen Paares
D: Kommentare, z. B., tailende Peaks, Basis-Linie-Drift, hoher Druck usw.
E: …
Selbstverständlich sollten nur solche Optimierungsparameter berücksichtigt werden, die brauchbare Ergebnisse geliefert haben, d. h. die Matrix sollte nach jedem Experiment aktualisiert werden. Ergibt sich beispielsweise mit dem Lösungsmittel „THF“ keine taugliche Trennung, so sollte Spalte 6 in Abbildung 2 gänzlich entfallen oder durch einen anderen Optimierungsparameter ersetzt werden. Gegebenenfalls kann für A bis D eine Gewichtung vorgenommen werden. Aber auch ohne Gewichtung kann ich auf einen Blick sehen, welche Säule-Eluent-Kombination beispielsweise meinen individuellen Kriterien genügt. Eine ähnlich aufgebaute Matrix
ermöglicht bei Robustheitsexperimenten einen schnellen Überblick über das Ausmaß von diversen Einflüssen, s. Abbildung 3:
Abb. 3 Zum Einfluss von Methodenparametern auf das Ergebnis (Methodenrobustheit), Erläuterungen, siehe Text
Auf der linken Seite der Matrix werden die chromatographischen Kenngrößen aufgelistet, deren Beeinflussung durch Variation von Methodenparametern untersucht werden soll. In der Zeile im unteren Teil der Matrix werden die vorgenommenen Veränderungen eingetragen, auch diese Zeile ist individuell zu gestalten: Sowohl was die zu untersuchenden Parameter betrifft (pH-Wert, Lösungsmittel,…), als auch das Ausmaß für die Veränderung (+/- 0,5 pH-Einheiten, X % B,…). Bei der Überprüfung der Laborpräzision (“intermediate precision“) beispielsweise könnte als Parameter „Anwender 2“, „Gerät 2“ usw. stehen. Die horizontalen Linien entsprechen dem sich jeweils ergebenden Zahlenwert (Fläche, Retentionszeit,…) bei den üblichen Trennbedingungen, also bei solchen, die nach erfolgter Methodenentwicklung festgelegt wurden. Die einzelnen Veränderungen werden durch schraffierte Flächen optisch dargestellt, der Maßstab kann ebenfalls in % oder als Zahlenwert individuell angelegt werden. Je nach Stärke der Veränderung kann anschließend entschieden werden, welche dieser Parameter als Systemeignungsparameter für den späteren Systemeignungstest in Frage kommen und welche Anforderungen (akzeptierte Bandbreite des jeweiligen Zahlenwertes) zu formulieren sind. Die beispielhafte Darstellung in Abbildung 3 wäre kurz wie folgt zu interpretieren: Diese Komponente reagiert zum einen sehr empfindlich auf eine Abnahme des pH-Wertes, die Trennung erfolgt offensichtlich in der Nähe des pKSBereichs. Dadurch nimmt die Retentionszeit mehr als die üblichen 10% für ähnliche Trennungen ab. Ebenso die Auflösung – hier wird offensichtlich ein benachbarter Peak schlecht abgetrennt. Darüber hinaus liegt eine pH-Wert-Abhängigkeit der UVAbsorption vor, vgl. Änderung der Peakfläche. Zum anderen ist scheinbar bei dieser Methode auch eine Zunahme der Temperatur kritisch (pH-Wert-Änderung durch
Temperatur-Änderung?). Was die anderen Parameter betrifft, scheint die Methode recht robust zu sein. Eine derartige Darstellung kann dem Validierungsbericht beigefügt werden. Schließlich erleichtert eine solche auch die Entscheidung im Falle einer späteren Änderung in der Methode: Gilt diese Änderung noch als „Justierung“ (Anforderung X erfüllt?) oder besteht tatsächlich Revalidierungsbedarf? Einen ökonomischen Weg, Optimierungs- und Robustheitsexperimente auf ein Minimum zu reduzieren, ermöglichen Softwarebasierte QbD-und DoE-Ansätze.
Extrakt: Vorgehensweise und „Philosophie“ des Konzepts
- Vorgehensweise
Zur Verfügung stehende Zeit: Eine Nacht und ein Tag - Säulenschaltventil mit sechs recht unterschiedlichen Säulen plus linearer Gradient (z. B. von 30% auf 90% ACN) bei drei (evtl. 4) pH- Werten, z. B. 2, 4,5 und 7 (evtl. 10)
- Temperatur +/- 30°C
- Gradient 20% auf 80% B und höherer Fluss sowie 30% auf 60% B und Erhöhung der Gradientendauer (Schritt 3 und 4 kombinierbar!)
Also: ∆ Säulen, ∆ pH-Wert, 2-3 lineare Gradienten, ∆ Temperatur
Zur Verfügung stehende Zeit: 2-3 Tage, zwei Nächte
Wie oben, zusätzlich: 50% ACN gegen MeOH bzw. 10% gegen THF ersetzen, mehr Gradienten-Experimente
Zur Verfügung stehende Zeit: Eine Woche
Wie oben, zusätzlich:
- Sechs/zwölf weitere Säulen werden während einer zweiten/dritten Nacht einem pH-Wert-Screening unterzogen
- gleicher pH-Wert allerdings eine andere Säure/Base
- mehr Gradienten-Experimente
- evtl. ∆ Säulendimensionierung
- Wenn möglich: Einsatz einer DAD-MS/MS-Kopplung
- Bei Bedarf(?!): Peakhomogenität mittels orthogonalen Tests sowie Säulenstabilität überprüfen
- Die „Philosophie“ dahinter: Variiere systematisch (relevante) Parameter und wende dabei das Prinzip der Ökonomie an. Das bedeutet:
- Orientierungsversuche: Verwende kurze Säulen, um schnell Trends zu erkennen
- Gehe ökonomisch und systematisch vor: Nutze die Nacht für die Fleiß-Arbeit und treffe Entscheidungen tagsüber
- „Wahrheit“ wichtig? Traue keinem Peak – sei er auch so schmal…
- Soll es eine zuverlässige Routinemethode werden? Denk´ früh genug an die Anforderungen des Alltags
Beratung zum Thema: „Begutachtung/Verbesserung von HPLC-Methoden“
Kurs zum Thema: „Optimierung in der HPLC – effizient und zielgerichtet“
Buch zum Thema: S. Kromidas (Hrsg.) „HPLC richtig optimiert“

