Unser Kunde sagte: „Ich brauche für meinen Kunden eine Methode mit einer guten Präzision und ohne Ausreißer – alles andere interessiert mich nicht.“ Dieser Kunde ist wichtig, hat keine Ahnung und wir wollen ihn zufrieden stellen. Wie machen wir das?“ Zusammenfassung: Gute Präzision? So klappt´s: Hervorragende Werte für die Präzision werden bekannterweise durch eine große Anzahl an Werten erreicht; ferner auch, wenn Messpräzision (Streuung der Werte bedingt lediglich durch das Gerät) statt Methodenpräzision ermittelt wird. Noch „krasser“: Keine Injektion aus sechs vials mit je einer Standardlösung, sondern sechs (drei) Wiederholinjektionen aus einem vial. Sind Ausreißer vorhanden? So sind keine zu befürchten: Es sollte als Ausreißertest natürlich der Dixon-Test verwendet werden a) Kleine Anzahl an Werten: Egal wie stark ein Wert abweicht, wird er kaum als Ausreißer zu deklarieren sein b) Starke Streuung der Werte – keine Probleme c) Große Anzahl an Werten (mehr als sechs); da der Dixon-Test hier ungeeignet ist, sind auch hier kaum Ausreißer zu „befürchten“ Die Story: Anfang des Jahres hat manch eine(r) gute Vorsätze für das neue Jahr; einige wollen vielleicht gar „bessere“ Menschen werden. Lasst uns dennoch für einen Moment ein raffiniertes, böses Teufelchen spielen und uns überlegen, wie man einen Unwissenden begeistern kann. Neben…
„Wir haben bei der Bestimmung vom LOQ starke Schwankungen. Auch an unterschiedlichen Geräten und mit neuen Säulen. Woran kann das liegen? Als LOQ-Kriterium haben wir wie üblich ein S/N-Verhältnis von 10:1.“ Antwort: Die Erfahrung zeigt, dass hier mit recht großen Integrationsfehlern zu rechnen ist. Wir konnten zeigen (s. Infos am Ende des Beitrages), dass kaum eine kommerzielle Software in der Lage ist, bei automatischer Integration und einem S/N-Verhältnis von 10:1 so zu integrieren, dass der Fehler – bei optimalen (!) Bedingungen (BL-Trennung, kaum Drift usw.) – nicht mindestens 5-10 % beträgt. Siehe dazu weiter unten die Werte in der Tabelle für den ersten, kleinen Peak (oben): In den grau schraffierten Feldern befinden sich Werte mit einer Abweichung von mehr als 1 % vom richtigen Wert, bedingt durch eine fehlerhafte Integration. Einige Kommentare und Erläuterungen zu den Werten der Tabelle: Die verwendeten Einstellparameter („Settings“) wie Dwell-Time, Time Constant, Sample Rate usw. können – vor allem bei kleinen Peaks – die Integration beeinflussen. So ist beispielsweise die Abweichung vom richtigen Wert beim ersten Peak bei einem Threshold-Wert von 50 12,50 %, bei einem Threshold-Wert von 100 17,78 % (EZChrom) Bei einigen Software-Programmen können unterschiedliche Integrationsalgorithmen angewandt werden. Die erhaltenen Ergebnisse können…
„Validierung ist aufwendig und teuer; was ist das Minimum an Validierung, was wir machen müssen?“ Zwei Vorbemerkungen Vor einer Antwort halten wir wie folgt fest: 1. Es gibt viele Definitionen zur Validierung, eine davon lautet: „Das Ziel bei der Validierung einer analytischen Methode ist zu zeigen, dass sie für den beabsichtigten Zweck geeignet ist.“ 2. Validierung ist – anders als z. B. GLP – kein Gesetz. Es gibt demnach de jure keine offizielle Stelle, die bzgl. Umfangs, Validierungstiefe, Durchführung, Revalidierungbedarfs etc. gesetzlich bindende Vorgaben macht. Somit jetzt schon einige Schlussfolgerungen: Validierung ist demnach etwas recht Individuelles: Um was geht es in einem aktuellen Fall eigentlich? Muss ich eher formale Sachen beachten, muss also eine wichtige Person/Organisation lediglich „nicken“? Oder stehen analytische Gesichtspunkte im Vordergrund, die notwendiger-/sinnvollerweise zu beachten sind? Sehr wohl ergibt sich häufig de facto aus bestimmten Zwängen/Gegebenheiten genau „was“, „wie“, und „wieviel“ an Validierung zu tun ist. Wenn ich diese Vorgaben missachte, bekomme ich beispielsweise keine Zulassung für mein Produkt bzw. kann ich besagte Methode gar nicht anwenden. Wenn ich solchen Zwängen nicht unterliege, kann ich selbst denken und dem „beabsichtigten Zweck“ gemäß handeln. Das heißt, ich suche aus der „Validierungsklaviatur“ (Richtigkeit, Präzision, Linearität, Robustheit etc.) diejenigen Validierungsparameter…
Im diesem Tipp haben wir uns mit verwirrenden Abkürzungen und nicht ganz glücklichen Synonymisierungen beschäftigt. Im hiesigen Tipp möchte ich auf vermeintlich eindeutige Begriffe und Angaben eingehen unter denen evtl. doch Unterschiedliches verstanden wird. Ich habe zwei Begriffe aus der HPLC und drei aus dem Bereich der Validierung ausgesucht.
Beispiele
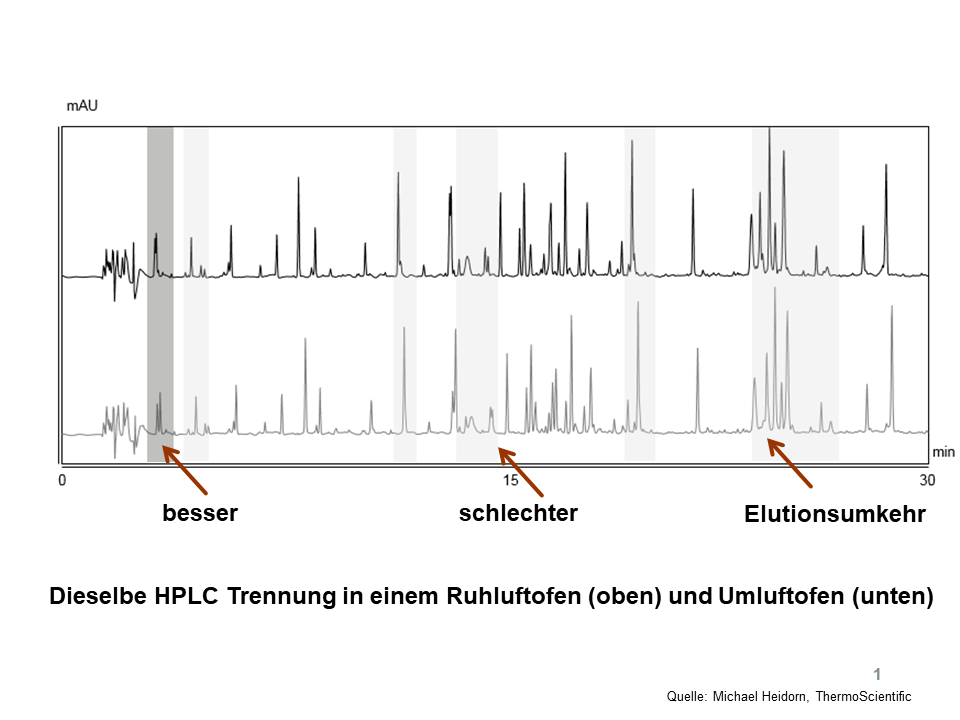
Luftofen
Zwei Labore möchten Ergebnisse vergleichen, die Hardware ist identisch, beide verwenden einen qualifizierten „Luftofen“. Sollte allerdings der eine ein Ruhluftofen und der andere ein Umluftofen (bzw. das gleiche Gerät aber im unterschiedlichen Modus betrieben) sein, könnten die Ergebnisse evtl. divergieren. Siehe dazu ein Beispiel in Abbildung 1: Bei gleicher Temperaturanzeige am Display kann an unterschiedlichen Luftöfen die Auflösung besser oder schlechter sein, auch Elutionsumkehr ist denkbar, weil ganz einfach die tatsächlich herrschende Temperatur in der Säule eine andere ist.
Abbildung 1. Unterschiedliche Ergebnisse mit unterschiedlichen Luftöfen, Details, siehe Text
Oberfläche des Materials in m2/g
In der Literatur und auch In Broschüren von Herstellern werden häufig die physikalisch-chemischen Daten der verwendeten bzw. angebotenen Materialien angegeben. Es kann leicht passieren, dass man übersieht, dass einmal die Rede von „effektive Oberfläche“ und einmal von „spezifische Oberfläche“ ist. Die Zahlenwerte sind nicht vergleichbar. So entspricht beispielsweise eine effektive Oberfläche von 200 m2/g einer spezifischen Oberfläche von 102 m2/g.
Signal/Rauschen-Verhältnis
Ein Zitat aus der amerikanischen Pharmakopöe:
“The signal-to-noise ratio (S/N) is a useful system suitability parameter. The S/N is calculated as follows: S/N = 2H/h”.
Als Bestimmungsgrenze wird oft ein Signal/Rauschen-Verhältnis von 10:1 verlangt/erwartet. Da jedoch die „2“ bereits in der Formel enthalten ist, bedeutet „10:1“: 20 mal die Peakhöhe zum Rauschen. In manch´ einem Labor wird allerdings als Bestimmungsgrenze tatsächlich 10 mal die Peakhöhe zum Rauschen genommen. Es liegt auf der Hand, dass Ergebnisse nicht vergleichbar sind, zumal die Integration in diesen Bereichen nicht unproblematisch ist…
Genauigkeit
Genauigkeit ist der Oberbegriff von Richtigkeit und Präzision; wenn ein Ergebnis frei ist von systematischen Fehlern, spricht man von einem richtigen Ergebnis. Wenn ein Ergebnis frei ist von zufälligen Fehlern, spricht man von einem präzisen Ergebnis. Wenn nun ein Ergebnis frei ist von systematischen und zufälligen Fehlern, also wenn jenes richtig und präzise ist, spricht man von einem genauen Ergebnis. Es ist allerdings so, dass oft „Genauigkeit“ gleich „Richtigkeit“ gesetzt wird. Die Konfusion wird nicht gerade geringer, als im Englischen für „Richtigkeit“ folgende drei Begriffe benutzt werden: „Accuracy“, „Accuracy of The Mean“, „Trueness“. Hier muss man mit den Gesprächspartnern früh genug für Klarheit der Begrifflichkeiten sorgen.
Linearität
Zwei Pharma-Labore möchten die Daten für die Linearität vergleichen. Die Geräte sind gleich, es werden Standards mit Placebo versetzt verwendet, man einigt sich auf neun unabhängige Werte. Nur: Das eine Labor wendet eine immer wieder anzutreffende Praxis im Pharma-Umfeld „3,3,3“, also drei Konzentrationen, a drei Bestimmungen. Das andere Labor verwendet tastsächlich neun unterschiedliche Konzentrationen und je eine Bestimmung. Auch hier sind die Ergebnisse nicht vergleichbar, auch dann wenn in beiden Fällen sich ein Korrelationskoeffizient mit drei Neuner nach dem Komma ergibt.
Der Ausweg heißt Kommunikation
Es ist sicherlich nicht einfach, derartige Missverständnisse gänzlich zu vermeiden. Man kann nur stets versuchen, eine enge und offene Kommunikation mit den Gesprächspartnern zu pflegen.
Von Mike Hillebrand, Aventis Der Fall Ein Baustein des Methodentransfers ist die „Intermediate Precision“ (Laborpräzision). Dabei werden in beiden Laboratorien, dem aufnehmenden und dem abgebenden, Vergleichsanalysen mit der validierten, zu transferierenden Methode durchgeführt. Diese sollen sicherstellen, dass das empfangende Laboratorium in der Lage ist, die neue implementierte Methode anzuwenden. Dadurch ist eine vollständige Validierung der Methode im empfangenden Laboratorium nicht nötig. Das Design der „Intermediate Precision“ richtet sich nach dem Analyseverfahren, welches es zu transferieren gilt. Beispielsweise wird für eine Gehaltsbestimmung häufig ein Design verwendet, bei dem zwei Mitarbeiter pro Labor zum Einsatz kommen. Ziel ist das Prüfen einer Charge….
Der Fall Einstellparameter wie Zeitkonstante („time constant“), Peakwidth, Datenrateaufnahme („sample rate“), Bandwidth etc. können die Integration und somit Peakfläche und Befund stark beeinflussen. Wir haben uns bereits darüber unterhalten, Details finden sich in (1), (2) und (3). In wie weit beeinflussen nun diese Parameter auch die Streuung der Werte im Falle von Wiederholmessungen? Die Lösung Nachdem ich in der Literatur diesbezüglich nicht fündig geworden bin, wurde in einer kleinen Studie an einer UPLC-Anlage der Einfluss diverser Einstellparameter auf den Vk systematisch untersucht. Dazu wurde zunächst mithilfe von Systemeignungstests die Zuverlässigkeit der Anlage überprüft. Anschließend wurden aus einem vial je 10…