Der Fall Im HPLC-Umfeld wird immer wieder die Frage diskutiert, ob denn für das Entgasen der mobilen Phase der eingebaute Degasser ausreicht. Ist in etwa ein zusätzliches Entgasen im Ultraschallbad sinnvoll/notwendig? Die Lösung Ich habe mich mit dieser Frage beschäftigt und dazu einige Experimente durchgeführt. Nachfolgend in verdichteter Form die Ergebnisse: Ein technisch ordnungsgemäß funktionierender Degasser reicht in der Regel vollkommen aus – vorausgesetzt, die Schläuche bzw. Membrane sind neu, denn: Mit der Zeit entstehen Mikrorisse, jene führen dazu, dass erstens kein gutes Vakuum erzeugt werden kann und dass zweitens Bestandteile der mobilen Phase an der dort größer gewordenen Oberfläche…
Liebe Kolleginnen, liebe Kollegen, vielleicht können Sie sich an die Fragen von Chromy an Peaky im HPLC-Tipp von Dezember erinnern. Hier sind noch einmal die Fragen sowie die Lösung: Chromy: Wir suchen zunächst ein Hauptwort, das hat neun Buchstaben. Zuerst musst du dieses Hauptwort raten. Dann musst du Wörter suchen, die senkrecht zu diesem Hauptwort angeordnet sind. Ich sage dir über welche Buchstaben des Hauptworts die einzelnen Wörter hinkommen. In diesen Wörtern sind Buchstaben des Hauptworts enthalten. Hier siehst du das Ganze. Schaffst du es in neun Minuten? Hauptwort: Das ist oft das Ziel Nummer Eins in der Chromatographie: Der…
Der HPLC-Tipp im Dezember Liebe Kolleginnen, liebe Kollegen, es ist eine Zeit her, dass wir den kleinen Peaky Säure und den großen Chromy von Hydrophobenhausen haben zu Wort kommen lassen. Nun ist es wieder soweit, horchen wir Ihnen einfach – nur wo sind sie denn? Klar doch: Peaky der schnelle und Chromy der langsame schlendern gemütlich durch die Säule. Peaky: Du Chromy, ich bin soooo froh, dass wir keine Maske tragen müssen. Unser Abstand zwischen uns ist soooo groß, da kann wirklich nichts passieren, juhu! (Peaky zeigt dem Corona-Virus frech die Zunge und hantiert noch an seine Nase herum). Chromy:…
Sprechen wir alle die gleiche „Sprache“? (III)
In diesem und in diesem Tipp haben wir uns über Probleme unterhalten, die dadurch entstehen, dass unter bestimmten Begriffen etwas Anderes verstanden wird. Oder aber auch, wenn ähnlich klingende Begriffe etwas Anderes bedeuten. Derartige Missverständnisse gibt es zur Genüge, deswegen greife ich das Thema noch einmal auf. Ich beschränke mich heute auf vier Beispiele in Regelwerken, also Infos, die an AnwenderInnen im regulierten Umfeld adressiert sind.
Systemeignungstest (System Suitability Test, SST)
In der USP steht: „The tests are based on the concept that the equipment, electronics, analytical operations, and samples analyzed constitute an integral system that can be evaluated as such”
Im gleichen Abschnitt etwas weiter steht allerdings auch: “These system suitability tests are performed by collecting data from replicate injections of standard or other solution as specified”
Diese, beide existierenden Aussagen führen dazu, dass in einem Labor für einen SST „samples“, also reale Proben, z. B. Wirkstoff plus Placebo, verwendet werden, in einem anderen Labor jedoch lediglich Standards. Die Ergebnisse sind dabei nicht immer vergleichbar, weil jene (Peakfläche, Peakform, Präzision) evtl. von der Matrix beeinflusst werden.
Und noch ein Detail in diesem Zusammenhang: In der JP ist der Text im Prinzip identisch mit dem Text in der USP (siehe weiter oben) jedoch mit dem Zusatz „operators“(!): „…and based on the concept that the equipments, electronic data processing systems, analytical operations, samples to be analyzed and operators constitute an integral system that can be evaluated“.
Temperatur
Gerade bzgl. „Temperatur“ gibt es in Regelwerken abweichende Definitionen, nachfolgend einige Beispiele zu „kalt“:
EP : Cold or cool: 8°C to 15°C
USP : Cold: Any temperature not exceeding 8°C
Cool: Any temperature between 8° and 15°C
JP : Cold: 1°C – 15°C
WHO: Cool: Store between 8°-15°C
Ferner: Der Terminus “Standard Temperature” wird nur von der JP (20°C) und „Ambient Temperature“ nur von der WHO (between 15° to 25°C) verwendet.
Nachfolgende Tabelle gibt einen Überblick über die Temperaturbereiche in einzelnen Regelwerken wider.
„In“ vs. „to“
In der Prüfvorschrift einer Monographiemethode steht: „…Disolve 100 g X to 1000 ml demin. water…“. Etwas weiter unten im Text steht: „…Disolve 2,8 g Y in 1000 ml demin. water and adjust to pH 4.5 with…“.
Verstehen alle AnwenderInnen dieser PV das gleiche bei diesen Angaben, wird die mobile Phase/die Probelösung auf der gleichen Art und Weise angesetzt, zumal es sich einmal um Feststoff und einmal um Flüssigkeit handeln kann?
„Relative Retention“ vs. „Relative Retention Time”
In der USP ist einmal die Rede von „Relative Retention (r)“ und einmal von „Relative Retention Time (RRT)“ („unadjusted relative retention“). Im ersten Fall handelt es sich um den Quotienten aus zwei Retentionszeiten, normiert um die Totzeit tM, r = tR2 tM/tR1 tM. Im zweiten Fall lediglich um den Quotienten aus zwei Retentionszeiten RRT = tR2/tR1 .
Diese ähnliche klingende Größen können Verwirrung stiften: Bei „Relative Retentionszeit“ bezieht man beispielsweise die Retentionszeit einer Verunreinigung auf die Retentionszeit der Hauptkomponente, um gerade diese Verunreinigung zu identifizieren. Dies ist zwar eine übliche Praxis (USP: “Comparisons in USP are normally made in terms of unadjusted relative retention”) jedoch problematisch, denn: Ich beziehe eine Variable (Retentionszeit der Nebenkomponente) auf eine andere Variable (Retentionszeit der Hauptkomponente), weil ja auch die Retentionszeit der Hauptkomponente sich durch Änderung der Eluentenzusammensetzung oder des pH-Wertes oder der Temperatur selbstverständlich ändern kann. Die Verwendung der relativen Retention (Retentions- oder Kapazitätsfaktor, k) macht aus chromatographischer Sicht mehr Sinn: Durch die Division mit der Totzeit, tM, normalisiere ich, weil ich mich auf eine Zeit beziehe – eben tM –, die definitionsgemäß von stationärer und mobiler Phase inkl. pH-Wert sowie Temperatur nicht beeinflusst wird. Ich beziehe eine Variable (Retentionszeit von X) auf eine Konstante (Totzeit, also Retentionszeit einer inerten Komponente), Ergebnis: Die relative Retention (Retentionsfaktor k) ist zur Identifizierung einer Komponente bei konstanten chromatographischen Bedingungen sicherer als die relative Retentionszeit.
Fazit:
Wenn es um Entscheidungen geht, steht oft sinnvollerweise die Vergleichbarkeit von Ergebnissen im Vordergrund, die „Richtigkeit“ von absoluten Zahlen ist häufig von untergeordneter Bedeutung, da Richtigkeit oft unbekannt. Um dies zu gewährleisten bedarf es der gleichen „Sprache“ und der exakt gleichen Anwendung von Tools und Größen.
Sprechen wir alle die gleiche „Sprache“? (II)
Im diesem Tipp haben wir uns mit verwirrenden Abkürzungen und nicht ganz glücklichen Synonymisierungen beschäftigt. Im hiesigen Tipp möchte ich auf vermeintlich eindeutige Begriffe und Angaben eingehen unter denen evtl. doch Unterschiedliches verstanden wird. Ich habe zwei Begriffe aus der HPLC und drei aus dem Bereich der Validierung ausgesucht.
Beispiele
Luftofen
Zwei Labore möchten Ergebnisse vergleichen, die Hardware ist identisch, beide verwenden einen qualifizierten „Luftofen“. Sollte allerdings der eine ein Ruhluftofen und der andere ein Umluftofen (bzw. das gleiche Gerät aber im unterschiedlichen Modus betrieben) sein, könnten die Ergebnisse evtl. divergieren. Siehe dazu ein Beispiel in Abbildung 1: Bei gleicher Temperaturanzeige am Display kann an unterschiedlichen Luftöfen die Auflösung besser oder schlechter sein, auch Elutionsumkehr ist denkbar, weil ganz einfach die tatsächlich herrschende Temperatur in der Säule eine andere ist.
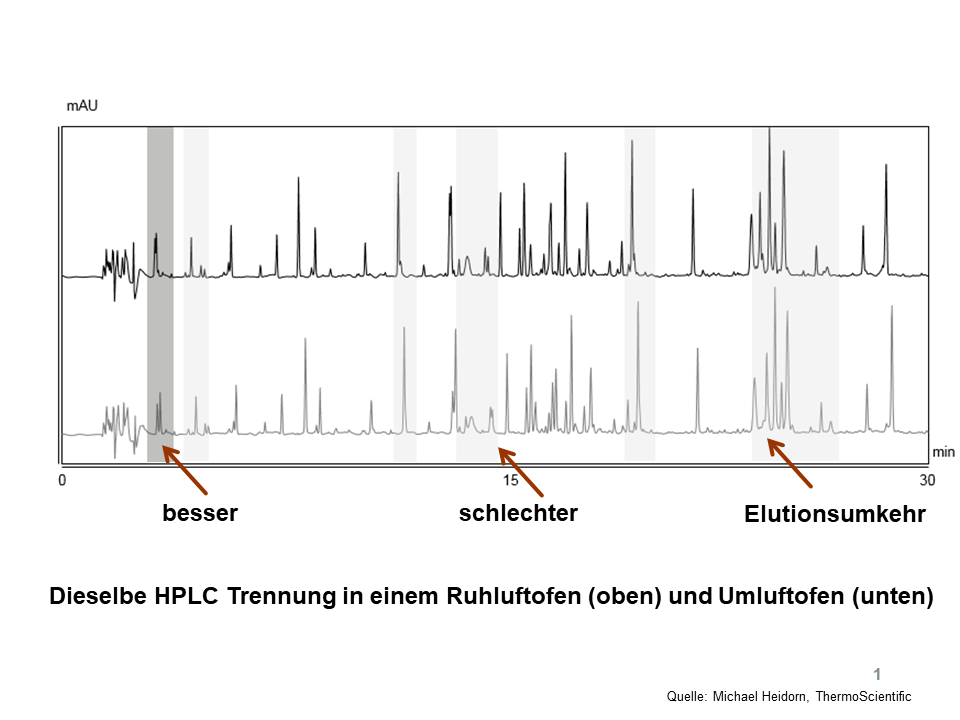
Abbildung 1. Unterschiedliche Ergebnisse mit unterschiedlichen Luftöfen, Details, siehe Text
Oberfläche des Materials in m2/g
In der Literatur und auch In Broschüren von Herstellern werden häufig die physikalisch-chemischen Daten der verwendeten bzw. angebotenen Materialien angegeben. Es kann leicht passieren, dass man übersieht, dass einmal die Rede von „effektive Oberfläche“ und einmal von „spezifische Oberfläche“ ist. Die Zahlenwerte sind nicht vergleichbar. So entspricht beispielsweise eine effektive Oberfläche von 200 m2/g einer spezifischen Oberfläche von 102 m2/g.
Signal/Rauschen-Verhältnis
Ein Zitat aus der amerikanischen Pharmakopöe:
“The signal-to-noise ratio (S/N) is a useful system suitability parameter. The S/N is calculated as follows: S/N = 2H/h”.
Als Bestimmungsgrenze wird oft ein Signal/Rauschen-Verhältnis von 10:1 verlangt/erwartet. Da jedoch die „2“ bereits in der Formel enthalten ist, bedeutet „10:1“: 20 mal die Peakhöhe zum Rauschen. In manch´ einem Labor wird allerdings als Bestimmungsgrenze tatsächlich 10 mal die Peakhöhe zum Rauschen genommen. Es liegt auf der Hand, dass Ergebnisse nicht vergleichbar sind, zumal die Integration in diesen Bereichen nicht unproblematisch ist…
Genauigkeit
Genauigkeit ist der Oberbegriff von Richtigkeit und Präzision; wenn ein Ergebnis frei ist von systematischen Fehlern, spricht man von einem richtigen Ergebnis. Wenn ein Ergebnis frei ist von zufälligen Fehlern, spricht man von einem präzisen Ergebnis. Wenn nun ein Ergebnis frei ist von systematischen und zufälligen Fehlern, also wenn jenes richtig und präzise ist, spricht man von einem genauen Ergebnis. Es ist allerdings so, dass oft „Genauigkeit“ gleich „Richtigkeit“ gesetzt wird. Die Konfusion wird nicht gerade geringer, als im Englischen für „Richtigkeit“ folgende drei Begriffe benutzt werden: „Accuracy“, „Accuracy of The Mean“, „Trueness“. Hier muss man mit den Gesprächspartnern früh genug für Klarheit der Begrifflichkeiten sorgen.
Linearität
Zwei Pharma-Labore möchten die Daten für die Linearität vergleichen. Die Geräte sind gleich, es werden Standards mit Placebo versetzt verwendet, man einigt sich auf neun unabhängige Werte. Nur: Das eine Labor wendet eine immer wieder anzutreffende Praxis im Pharma-Umfeld „3,3,3“, also drei Konzentrationen, a drei Bestimmungen. Das andere Labor verwendet tastsächlich neun unterschiedliche Konzentrationen und je eine Bestimmung. Auch hier sind die Ergebnisse nicht vergleichbar, auch dann wenn in beiden Fällen sich ein Korrelationskoeffizient mit drei Neuner nach dem Komma ergibt.
Der Ausweg heißt Kommunikation
Es ist sicherlich nicht einfach, derartige Missverständnisse gänzlich zu vermeiden. Man kann nur stets versuchen, eine enge und offene Kommunikation mit den Gesprächspartnern zu pflegen.
Der Fall
Die unterschiedliche Bedeutung von Hersteller-Abkürzungen sowie die Synonymisierung von an für sich unterschiedlichen Begriffen führen immer wieder zur Konfusion. Nachfolgend möchte ich dazu einige typische Beispiele anführen. Ferner: Unterschiedliche Interpretation von chromatographischen Begriffen und Formulierungen wie auch das Gleichsetzen ähnlich klingender Begriffe kann zu unterschiedlichen Schlussfolgerungen führen. Auch ein Datenvergleich kann sich dadurch als problematisch erweisen. Mit dieser verwandten Problematik werden wir uns im diesem Tipp beschäftigen. Hier finden Sie einige Synonyme in der HPLC.
Die Lösung
Gleiche Abkürzungen
Fangen wir mit dem Einfacheren an: Gehen Sie bitte nicht davon aus, dass gleiche Abkürzungen stets das Gleiche bedeuten. Dazu einige Beispiele:
„HD“ bei Agilent: High Definition
„HD“ bei Macherey Nagel: High Density (stationäre Phase mit ca. 20% Kohlenstoff)
„SB“ bei Waters: Strong Bond
„SB“ bei Agilent: Stable Bond (sterischer Schutz, im Sauren stabil)
„HT“ bei Agilent und Thermo beispielsweise wird für „High Throughput“ verwendet, während bei Waters, Tosoh Bioscience und anderen „HT“ für „High Temperature“ steht.
Auch Zahlen können beim Namen einer Säule etwas Unterschiedliches bedeuten, hier einige Beispiele: Die „2“ ist oft der Hinweis auf ein endcappedes Material (z. B. Inertsil 2, LUNA 2). Bei „HT2“ jedoch (Tosoh Bioscience) ist die „2“ der Hinweis, dass jenes GPC-Material bis 220 °C stabil ist, während die üblichen Hochtemperatur-GPC-Materialien („HT“) nur bis 140°C-150°C stabil sind. Die „3“ bei Inertsil ODS 3 wird als Hinweis verwendet, dass es sich dabei um ein weiteres Produkt der Inertsil-Familie handelt, das Material soll eindeutig von Inertsil ODS 2 abgrenzbar sein, welches recht andere Eigenschaften aufweist. Eine „3“ allerdings nach einem „T“ (z. B. Atlantis T3, Waters) bedeutet, dass der Ligand bei diesem Material über eine trivalente Bindung mit der Oberfläche des Kieselgels verbunden ist (T: Erster Buchstabe aus dem Griechischen „tria“ = drei).
„RP“ bedeutet bekannterweise „Reversed Phase“. „RP“ allerdings direkt nach „C18“, also in etwa „Materialname C18 RP“, ist oft der Hinweis, dass sich auf der Oberfläche dieser C18-Phase zusätzliche polare Gruppierungen befinden, dass also jene stationäre Phase einen (zusätzlichen) polaren Charakter aufweist.
Es gibt erfreulicherweise auch einfache Fälle: Die Bedeutung einer Abkürzung ergibt sich zwangsläufig aus dem Zusammenhang: „ECD“ in der GC bedeutet Electron Capture Detector, in der HPLC, Electrochemical Detector.
Unglückliche Synonymisierungen
- Aus einem Buch „…Totvolumen, auch Verweilvolumen genannt…“ Diese Gleichsetzung ist problematisch, denn: Totvolumen (Dead Volume, Dispersionsvolumen) ist das Volumen der Apparatur vom Probengeber bis einschließlich Detektor ohne Säule. Ändert sich jenes, ändert sich die Peakform vor allem bei früh eluierenden Peaks, die Retentionszeit ändert sich dabei nur gering. Das Verweilvolumen (Verzögerungsvolumen, Dwell- oder Delay Volume) bei Gradientenanlagen ist das Volumen vom Mischventil bis zum Säulenkopf. Ändert sich jenes, kann eine Vielzahl von Effekten auftreten: Keine Änderung, Änderung der Retentionszeit, der Peakform, der Auflösung, mitunter immer wieder auch der Elutionsreihenfolge.
- Ebenfalls aus einem Text: „…Selektivität oder Spezifität …“. Diese Gleichsetzung ist nicht richtig: Selektivität ist die Fähigkeit einer Methode alle denkbaren Komponenten ohne gegenseitige Störung zu trennen. Spezifität ist die Fähigkeit einer Methode, eine Substanz oder eine Substanzklasse ohne Störung durch andere Komponenten zu trennen. Leider ist in den 1990er Jahren den Verfassern der Richtlinien der ICH (International Conference of Harmonisation) ein semantischer Fehler unterlaufen, sie verwendeten nämlich den Begriff „Specifity“. Da in diesem Papier (stillschweigend) chromatographische Methoden gemeint sind, sollte richtigerweise von „Selectivity“ die Rede sein, denn: Man schätzt sich in der HPLC-Welt glücklich, eine selektive chromatographische Methode entwickelt zu haben. Spezifität in der Chromatographie ist allenfalls nur theoretisch denkbar. Dieses sprachliche Missgeschick hat im Pharma-Umfeld manche Verwirrung gestiftet.
Das Fazit
Bei einer falsch verwendeten Synonymiesierung ist es zugegebenerweise schwierig dahinter zu kommen. Bei nicht 100%ig geläufigen Abkürzungen lohnt es sich bei Bedarf folgendes zu tun: Geschwind bei der Homepage des Herstellers nachschauen, was wohl damit gemeint ist. Es sei denn, allen Beteiligten ist klar, was mit der Abkürzung gemeint ist, z. B. bei HPLC doch eindeutig: High Pleasure Liquid Chromatography…
- Säurezusatz im Eluenten
- Gefahr für Niederschläge
- Vorsäule ja, aber was für eine?
Alternativen zu TFA
Trifluoressigsäure (TFA) wird gerne zum Ansäuern in der RP-HPLC verwendet, häufig bei – eventuell erst in der Zukunft geplanten – LC-MS-Kopplungen. TFA bereitet jedoch bekanntlich einige Probleme, so in etwa Basisliniendrift, immer wieder Empfindlichkeitsverlust, TFA kann lange auf der Säule bleiben usw. Welche Alternativen hätten wir? Wenn Ameisensäure für bestimmte Trennungen nicht sauer genug ist, könnte man an Pikrin- oder an Sulfamin- oder an Difluoressigsäure (DFA) denken. Ferner – sollte ein Ionenpaarreagenz benötigt werden – an Methansulfonsäure. Noch ein Wort zu Phosphatpuffer: Phosphorsäure bzw. ein Phosphatpuffer bewährt sich seit langem in der RP-HPLC mit UV-Detektion. Sollten Sie mit der Trennung bzgl. Selektivität/Peakform bei Anwendung von Phosphorsäure oder Phosphatpuffer zufrieden sein, könnte man getrost eine LC-MS-Trennung wagen: Bei einer Verwendung von ca. 10 mM Phosphatpuffer müsste man erst nach 4-5 Stunden das Interface reinigen. Merke in diesem Zusammenhang folgende generelle Regel: Je ähnlicher der pH-Wert des Eluenten zum pKS-Wert des verwendeten Puffers ist, desto niedriger kann die notwendige Pufferkonzentration sein. Dennoch gilt: Das Dilemma gutes chromatographisches Ergebnis vs. Reinigungs-Aufwand kann nur individuell gelöst werden.
Niederschlag
Eine Verstopfung im Gerät durch einen Niederschlag ist immer ärgerlich. Es liegt auf der Hand, dass diese Gefahr mit steigender Puffer- und Acetonitril-Konzentration sowie bei niedrigen Temperaturen zunimmt. Nachfolgend einige Hinweise:
- Ab ca. 85% Acetonitril in der mobilen Phase und ≥ ca. 20 mM Puffer nimmt das Risiko von Niederschlag im Falle von dünnen, ≤ ca. 0,13 mm Kapillaren stark zu
- „Phosphatpuffer“ – nur welcher? K2HPO4 (Pufferbereich: pH-Wert = 6,5-7,5) macht kaum Probleme, die Löslichkeit ist sehr gut. Na2HPO4 dagegen, insbesondere bei hohem ACN-Anteil und ≤ ca. 25 °C, kann definitiv Probleme bereiten, denn: Die Differenz der Löslichkeit der zwei Salze beträgt mehr als Faktor 20! Im sauren gibt es generell kaum Schwierigkeiten
- Ammoniumacetat bei ≥ ca. 60% ACN: Farblose Kristalle, die beispielsweise in der Mischkammer ausfallen
Die „geeignete“ Vorsäule
Oft wird eine Vorsäule zum Schutz der Hauptsäule eingesetzt. Das Material der Vorsäule muss nicht unbedingt identisch mit dem der Trennsäule sein, es ist Regel-konform, wenn es z. B. auch „C18“ ist. Warum nicht identisch? Die Vorsäule hat in der Regel die Aufgabe, ziemlich „viel“ von der störenden Matrix zu adsorbieren, dabei soll sie nach Möglichkeit wenig Druck aufbauen. Diese Anforderungen führen zu folgenden sinnvollen Charakteristika für eine Vorsäule:
- Gute Beladbarkeit: Dazu soll das Material der Vorsäule eine möglichst große spezifische Oberfläche aufweisen, z. B ≥ 250 m2/g
- Große Bindungskapazität: Die Beladungsdichte des Materials sollte über 3 mMol/m2 der Kohlenstoffgehalt über ca. 18-20% C betragen
- Im Falle von Gradiententrennungen spielt die Teilchengröße eine untergeordnete Rolle. Somit kann die Teilchengröße in der Vorsäule ruhig 5 µm betragen, auch dann, wenn die analytische Säule 3 µm-Teilchen oder kleiner enthält. 5 µm- Teilchen sind haltbarer als 3 µm und bauen einen geringeren Druck auf.
Wird eine Vorsäule nicht zum Schutz der Hauptsäule, sondern zu einer Verbesserung der Trennung von sehr polaren Komponenten – also Elution solcher um oder kurz nach der Totzeit – eingesetzt, sollte das Material natürlich polarer als jenes der Hauptsäule sein. Im Falle einer C18-Hauptsäule kämen somit folgende Phasen für die Vorsäule in Frage:
CN, Phenyl-Hexyl, PFP, Hypercarb, Kieselgel oder Mixed-Mode.
Permanent auftretender Geisterpeak Faustregeln zu Log P und Log D Abnahme der Auflösung – eine mögliche Ursache Seit kurzem erscheint ein Geisterpeak bzw. stets ein Blindwert im Blank… Seit einiger Zeit taugt bei einer Methode – oder bei mehreren – die noch vor kurzem keine Probleme machte(n), ein Geisterpeak auf. Oder aber, Sie haben in Ihrem Blank auf einmal stets einen Blindwert. Und dieses Problem haben Sie unabhängig von der Anlage, dem/der Anwender(in) und sogar der Methode. D.h. das übliche Fehler-Ausschluss-Programm haben Sie gründlich aber leider erfolglos absolviert. Versuchen Sie in einem solchen Fall im Team sich zu erinnern,…
In diesem HPLC-Tipp geht es um diverse Probleme, die mit den Autosampler-Vials zu tun haben.
Vials, immer wieder vials…
- Beim Schütteln eines vials (bei mir leider immer wieder eine spontane Bewegung…) kann sich auf der Unterseite des vial-caps ein Substanzfilm bilden – die Reproduzierbarkeit der Injektion lässt zu wünschen übrig. Beim Vortexieren bzw. automatischen Schütteln ist diese Gefahr eher selten gegeben, die Präzision bei Wiederholinjektionen ist in der Regel besser
- Der Greifarm des Autosamplers hat evtl. ein vial verloren; wenn man Pech hat, versteckt sich jenes unter dem Karussell. Dadurch ist das Karussell nun etwas schief, die vials dort liegen ein wenig niedriger/höher, Ergebnis: Geänderte Peakflächen. Der Grund: Die Nadel stößt evtl. an den Wänden vom vial an oder sie kommt auf dem Boden auf. Oder aber im Falle von inhomogenen Probelösungen ergibt sich eine unterschiedliche Probekonzentration oder sogar ein Konzentrationsgradient . Und je nachdem, aus welcher Höhe die Nadel Probelösung ansaugt, ergeben sich Schwankungen der Peakfläche oder ein Trend dergleichen. Die Höhe eines vials kann sich auch dann verändern, wenn sich unter dem vial Schmutz, Dichtungsabrieb, Salzkriställchen usw. eingefunden haben
- Hellrote vs. dunkelrote vs. Silikon vs. vorgeschlitzte vs. „Sandwitch-Septen“; Unterschiede bezüglich Abriebs bzw. Verdampfens von Probelösungsmittel bzw. Neigung zu Memoryeffekt
- Vial richtig dicht vs. einfach „klack“ und somit eher locker aufgesetzt: Im ersten Fall ist die erste Injektion evtl. fehlerbehaftet (durch den Unterdruck beim erstmaligen Stechen der Nadel drückt sich etwas von der Probelösung in die Nadel hoch) im zweiten Fall sind alle Injektionen OK – auch die erste
- Vial, beispielsweise Nr. 18, geht immer wieder kaputt; das ist zwar ein seltener Fall, dennoch möchte ich ihn erwähnen: Durch einen Fabrikationsfehler kann beim 6-Port-Ventil passieren, dass die zwei Scheiben nicht 100% übereinander liegen. Bei wiederholten Problemen mit Injektionen nur aus einem bestimmten vial am besten zeitnah den Hersteller wegen eines Austausches kontaktieren
- Probleme beim Methodentransfer
Beim Methodentransfer tauchen oft deswegen Probleme auf, weil nicht alle Informationen ausgetauscht bzw. Begriffe unterschiedlich verstanden werden. Nachfolgend drei Beispiele betreffend die vials:
- „Wir arbeiten bei diesen geringen Volumina mit Aufsätzen, die sollt ihr auch verwenden“. „OK“. Nur: In einem Labor werden die beweglichen (und billigen) Aufsätze verwendet, im anderen die festen Aufsätze. Es ergeben sich womöglich unterschiedliche Peakflächen, weil im ersten Fall die Nadel links oder rechts die Vial-Wand berühren kann…
- „Wirkstoff X bleibt gerne hängen, ihr sollt vials mit inerter Oberfläche verwenden.“ „OK“. Nur: Versteht jede(r) unter „inert“ das Gleiche?
- Es werden statt Glas, PP-vials verwendet; die einen haben die normalen Eppi´s die anderen die low-bind Eppi´s …
- Die vials werden mit HCL behandelt; die Silanolgruppen auf der Glasoberfläche liegen undissoziiert vor, basische Wirkstoffe werden zwar nicht adsorbiert, Wasserstoffbrückenbindungen wären jedoch möglich
- Es werden silanisierte vials verwendet; analog einer endcappeden C18-Phase werden nicht nur keine basische sondern überhaupt keine polare Komponenten adsorbiert
- Es werden silikonisierte vials verwendet; durch die Silikon-Schutzschicht wird nicht lediglich Adsorption sondern auch eine Benetzung der Oberfläche verhindert, die Probelösung perlt einfach ab
- „Wir arbeiten mit vials mit so ´ne Verjüngung, weißt Du? „Ja, wir auch!“ Nur: Erstens, können solche Aufsätze unterschiedliche Länge aufweisen. Und zweitens kann der Durchmesser am Ende der Verjüngung unterschiedlich groß sein. Dadurch kann durch die Oberflächenspannung genau dort ein Luftbläschen entstehen – und dies abhängig vom Probelösungsmittel! Ergebnis: Unterschiedliche Peakflächen
In Abbildung 1 werden Aufsätze der Firma VWR gezeigt, die die hier
erwähnten Unterschiede demonstrieren.

Abbildung 1: Aufsätze für HPLC-vials unterschiedlicher Länge und unterschiedlicher Verjüngung, Details, siehe Text
Der Fall Sie arbeiten in einem regulierten Bereich und sind an strenge Prüfvorschriften gebunden. In einer solchen lautet die Forderung: „Auflösung R größer 1,5“, aber gerade diesen Wert erreichen Sie aktuell nicht. Welche regel-konforme Handgriffe kämen in Frage? Die Lösung Was möglich ist, hängt letzten Endes davon ab, wie genau die Angaben in der betreffenden PV sind. Ist in der Tat restlos alles – von der Eluentenzusammensetzung bis zu den Einstellungen („Settings“) – vorgegeben, so können Sie de facto es nur mit einer neuen Säule versuchen. Ist die PV etwas „weicher“, d.h. es sind nur die wichtigsten Parameter wie Säule,…

