Zusammenfassung: Orthogonaler Text: Verwende eine „ganz“ andere Säule (z. B. statt einer C18 nun eine PFP oder eine Mixed Mode) und/oder einen anderen Eluenten (mobile Phase statt mit ACN nun mit MeOH) und injiziere erneut. Ähnliche Substanzen gehen wahrscheinlich (etwas) andere Wechselwirkungen mit der stationären Phase ein. Somit offenbart sich, dass ein symmetrischer Peak evtl. doch nicht homogen ist. Der Fall In den letzten zwei HPLC-Tipps haben wir folgendes gesehen: Eine Änderung von Einstellparametern („Settings“) sowie „Manipulationen“ der Probelösung stellen schnelle Möglichkeiten dar, die Peakhomogenität zu prüfen. Heute geht es um den orthogonalen Test. Was ist das und was „bringt“ er? Die Lösung Am Ende einer Methodenentwicklung kommt häufig die Frage auf: „Habe ich alle Peaks trennen können, oder liegt womöglich irgendwo im Chromatogramm doch eine Koelution vor“? Jetzt kommt der orthogonale Test ins Spiel – die Idee dahinter: Man verwende eine völlig andere stationäre Phase oder einen anderen Eluenten und injiziert erneut. Es ist ziemlich unwahrscheinlich, dass zwei oder drei Komponenten bei Verwendung zweier gänzlich (!) unterschiedlichen Säulen bzw. Eluenten in beiden Fällen völlig gleich starke Wechselwirkungen mit der stationären Phase eingehen. Wenn nun mit einem Eluenten an zwei unterschiedlichen Säulen oder mit zwei unterschiedlichen Eluenten an einer Säule…
Der Fall Im HPLC-Tipp vom letzten Dezember war Peaky am Zweifeln, ob er es wirklich ist: „Bin ich überhaupt der Peaky? Und wenn ja, wieso fühlt es sich so an, als wäre ich mehrere …“ Es geht also um die Frage, ob ein Peak homogen ist oder womöglich doch eine Koelution vorliegt. Im vorliegenden HPLC-Tipp werden wir uns einfache Tests anschauen, die recht schnell durchzuführen sind: Keine Änderung der chromatographischen Parameter, keine Änderung der Apparatur. In den nächsten HPLC-Tipps werden wir uns sukzessiv mit aufwendigeren Methoden beschäftigen. Die Lösung Eine Veränderung von Einstellparameter („Settings“) kann helfen, innerhalb von Sekunden/Minuten die Peakhomogenität zu überprüfen. Es lohnt sich (auch) an solche „banale“, schnelle Möglichkeiten zu denken: Injektion bei einer anderen (niedrigeren) Wellenlänge, siehe Abbildung 1: Bei 271 nm erscheint der dritte Peak als ein Peak (unteres Chromatogramm), bei 225 nm sind zwei Peaks zu erkennen (oberes Chromatogramm) 2. Eine große Zeitkonstante („Filter Response“, „Filter Time Constant“) führt zwar zu einer ruhigeren Basislinie, die Peakbreite nimmt allerdings zu, man verliert an Auflösung. Folgendes Zahlenbeispiel aus einer „Technical Note“ von Waters, die freundlicherweise von Sascha Schifrin zur Verfügung gestellt wurde: Kein digitales Filtern: Auflösung (Resolution) 3,16, Peakkapazität 16 Zeitkonstante, 0,5 Min: Auflösung 1,82,…
Der Einfluss von Metallionen in der HPLC ist schon seit Längerem bekannt; sprechen wir eher von einem zwar vorhandenen, jedoch bis dato bei vielen Anwender:innen weniger sich stark im Focus befindenden Problem. In den letzten Jahren erlebt dieses Thema allerdings aufgrund der zunehmenden Wichtigkeit von Biomolekülen eine gewisse Renaissance. Um welche Metallionen handelt es sich? Wo kommen sie her und was bewirken sie? Sind sie vermeidbar? Und: Können sie entfernt werden? Doch bevor wir uns dem Thema widmen, zunächst ein kleiner semantischer Exkurs: Konfusion von Begrifflichkeiten Begriffe im hiesigen Zusammenhang werden nicht von allen Beteiligten einheitlich verwendet. Dadurch entsteht eine gewisse Konfusion. Nachfolgend eine kurze Einordnung, wobei diese weder den Anspruch auf Vollständigkeit erhebt, noch kann ich erwarten, dass alle mit dieser 100 % d’accord gehen. Bioinert; geringe Wechselwirkungen mit Hardware-Komponenten, geringes Risiko für carryover; ältester Begriff, bereits 1933 verwendet: Aluminium-Oberfläche als Alternative zu Eisen. Im Umfeld der HPLC sind die verwendeten Materialien hier Titan, Keramik, PEEK Biokompatibel; keine Korrosionsgefahr trotz Chlorid-Ionen im Eluenten; oft synonym bzw. gleich zu setzen mit Bio-LC und Stainless steel-free; verwendete Materialien: Titan und spezielle Legierungen, z. B. MP35N Metall-frei; verwendete Materialien: PEEK, Saphir Weitere anzutreffende Attribute sind: „Inert“, „truly bioinert“ (PEEK), schließlich „fully biocampatible“….
Der Fall Sie konnten für bestimmte Probleme (Geisterpeaks, Tailing etc.) das/die vial(s) inkl. Inhaltes als Ursache identifizieren. Und dies obwohl an der Methode eigentlich „nichts“ geändert wurde. An was sollten Sie – auch – denken? Die Lösung Probleme können zunächst durch die Injektion selbst hervorgerufen sein. So kann beispielsweise die Injektionsnadel durch ein Septumpartikelchen oder Salzkriställchen teilweise verstopft sein; oder die Nadelspitze ist aufgrund eines harten (dunkelroten) Septums minimal verbogen; oder sind die Purgeflüssigkeit und/oder die Lösung im Waschvial schlicht alt; oder schließlich verhindert Luft in der Spritze/Injektionsnadel das Ansaugen des eingestellten Injektionsvolumens. Hier wollen wir uns jedoch nur auf das/die vial(s) bzw. sein Inhalt als Fehlerquelle konzentrieren. Nachfolgend sind einige Ursachen aufgeführt, die zu einem veränderten Chromatogramm führen können: Veränderung der Peakfläche bzw. Peakform, zusätzliche Peaks, Retentionszeitverschiebungen etc. Beispiele für Veränderungen der Probelösung Der pH-Wert Ihrer wässrigen Probelösung hat sich durch Silanolgruppen an der Oberfläche des vials geändert, diese Änderung kann innerhalb einer Stunde über eine pH-Wert-Einheit ausmachen. Es kann sein, dass bei einer langen Sequenz sich eine größere – da zeitabhängige – pH-Wert-Veränderung bei den letzten vials stärker bemerkbar macht; oder Sie haben eine neue Charge von vials eingesetzt deren Oberfläche acidere Silanolgruppen aufweist; oder die Probe bleibt…
Typischen Symptomen können bestimmten Ursachen zugeordnet werden; oder aber die Zahl der infrage kommenden Ursachen kann durch Beurteilung der Symptome („Welche Veränderung kann dieses Symptom bedingen und welche definitiv nicht?“) wenigstens eingegrenzt werden. Nachfolgend werden beispielhaft sieben häufige „Symptome-Ursachen“ in der Routine-HPLC vorgestellt. Bei Bedarf erfolgt auch ein Kommentar. Für die vorgestellten Fälle gelten folgende Annahmen: Keine bewusste Änderung seitens der Anwender:innen, z. B. es wurde keine längere Säule eingebaut Es handelt sich vorwiegend um RP-Trennungen mit einem konzentrationsempfindlichen Detektor wie beispielsweise UVVis, Diodenarray, Fluoreszenz- oder Brechungsindexdetektor Weiter unten sind die häufigsten Ursachen genannt Symbole: ∆: Änderung H: Peakhöhe A:…
Der Fall Zu Beginn einer längeren Sequenz läuft alles bestens. Gegen Sequenz-Ende jedoch tauchen verstärkt Probleme auf, z. B. Geisterpeaks, Veränderung der Peakform und/oder der Peakfläche und nicht zuletzt Verschiebung der Retentionszeit. Welche Ursachen kommen in Frage? Die Lösung Es handelt sich hierbei wohl um Ursachen, die zeitabhängig sind. Nachfolgend eine Auswahl von Symptomen und möglichen Ursachen: Temperatur zum Ersten: Druck-, evtl. auch Peakfläche-Schwankungen Es finden manchmal aufgrund einer niedrigen Temperatur im Probengeber Nachfällungen statt, die erst später einsetzen und zu folgenden Problemen führen können: Verstopfung der Injektionsnadel, Niederschlag am Siebchen direkt am Säulenkopf usw. Vorgehen, um ein kommendes Problem…
Geisterpeaks – aber erst „später“ Verbesserung der Empfindlichkeit Geisterpeaks, die „später“ oder erst in einigen Tagen erscheinen Geisterpeaks erscheinen meist plötzlich. Man wundert sich wieso, hat man doch an den chromatographischen Bedingungen nichts geändert. Nun, es sind schon einige Situationen denkbar, in denen Geisterpeaks erst nach einer gewissen Zeit zu sehen sind. Nachfolgend einige Beispiele dazu (zur Problematik von Geisterpeaks ab und an siehe auch diesen Tipp): Katalytische Wirkung des Kieselgels Kieselgel ist ein guter Feststoffkatalysator; bei längeren Läufen werden womöglich nur solche Substanzen verändert, die lange an der Oberfläche des Materials haften und somit…
Der Fall Sie arbeiten in einem regulierten Bereich und sind an strenge Prüfvorschriften gebunden. In einer solchen lautet die Forderung: „Auflösung R größer 1,5“, aber gerade diesen Wert erreichen Sie aktuell nicht. Welche regel-konforme Handgriffe kämen in Frage? Die Lösung Was möglich ist, hängt letzten Endes davon ab, wie genau die Angaben in der betreffenden PV sind. Ist in der Tat restlos alles – von der Eluentenzusammensetzung bis zu den Einstellungen („Settings“) – vorgegeben, so können Sie de facto es nur mit einer neuen Säule versuchen. Ist die PV etwas „weicher“, d.h. es sind nur die wichtigsten Parameter wie Säule,…
In den letzten Jahren wurde eine Reihe moderner C18- sowie polarer RP-Phasen eingeführt. Da wären beispielhaft zu nennen: Chemisch geschützte („embedded“ phases), hydrophil endcappede sowie Mixed Mode Phasen und was die Matrix betrifft: Hybrid-, Core Shell- oder monolithische Phasen. Diese Materialien weisen vielfach Vorteile auf. Heißt es nun, dass bei der Entwicklung einer neuen Methode der Einsatz einer solchen modernen Phase die richtige Wahl wäre? Sollte man also nicht-endcappede Phasen als eine alte Technologie „ad acta“ legen?
Die Lösung
Nein. Für die Trennung von Substanzen mit ähnlicher Hydrophobie, aber mit Unterschieden in der Anordnung von Substituenten am Molekül oder von Doppelbindungen in einer Seitenkette (α, β-Isomerie, Stellungsisomerie) oder stark polare Komponenten sind Restsilanolgruppen für die Selektivität sehr wichtig. Dies wird an drei Beispielen demonstriert:
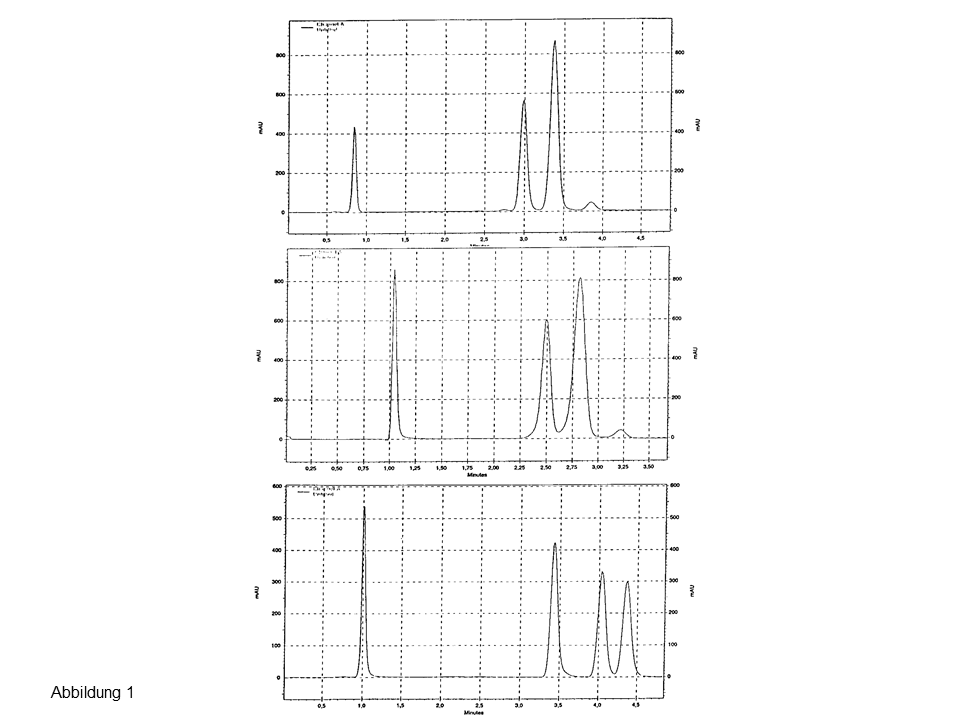
Beispiel 1: Trennung von Steroiden
Abb. 1: Trennung von drei Steroiden an zwei endcappeden (oben, Mitte) und an einer nicht endcappeden C18-Phase (unten), Erläuterungen siehe Text.
Das obere und mittlere Chromatogramm zeigen die Injektion von drei Steroiden (α, β-Isomere) an zwei modernen, hydrophoben Phasen. Steroid Nr. 2 und 3 koeluieren. Die Trennung gelingt an Resolve C18, einem älteren, nicht endcappeden Material, siehe unteres Chromatogramm in Abbildung 1.
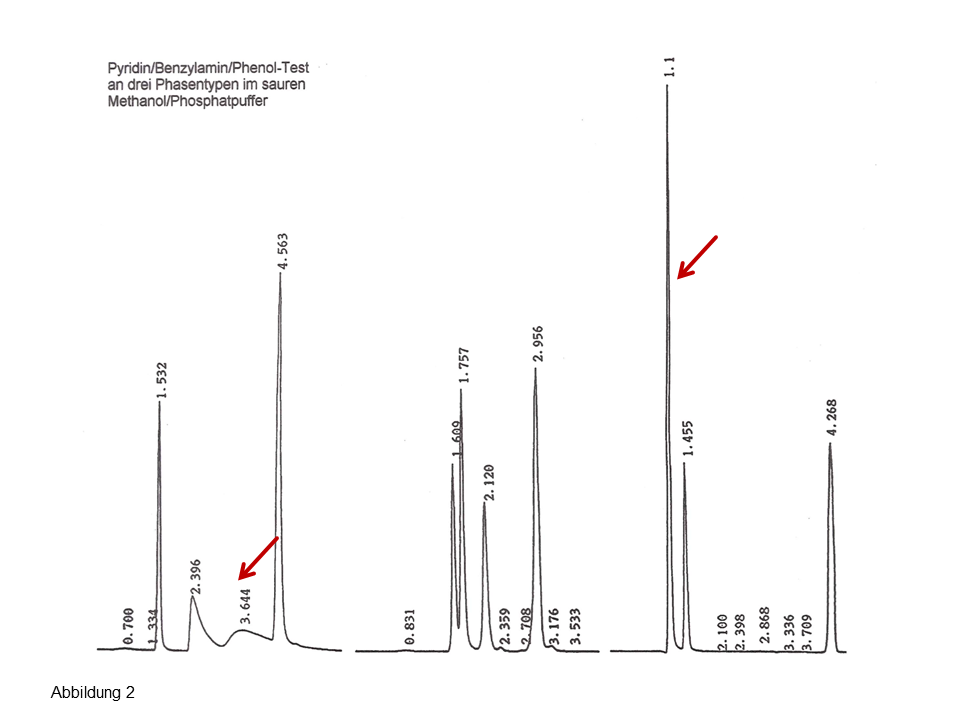
Beispiel 2: Trennung von starken Basen
Abb. 2: Injektion einer Mischung von drei polaren Komponenten auf eine silanophile (links) und eine hydrophobe, endcappede C18-Phase (rechts), Erläuterungen, siehe Text.
Auf der rechten Seite der Abbildung 2 wird die Injektion von Uracil (inerte Komponente), Pyridin, Benzylamin und Phenol an einer modernen endcappeden C18-Säule gezeigt. Die zwei Basen koeluieren (erster Peak), was vollkommen nachvollziehbar ist: Man kann nicht erwarten, dass eine hydrophobe, gründlich endcappede Phase eine gute polare Selektivität aufweist. Und das kann zu falschen Schlussfolgerungen führen: Eine gute Peaksymmetrie suggeriert im Routinealltag eine gute Selektivität… Das linke Chromatogramm zeigt die Injektion auf eine „alte“, stark silanophile Phase, Hypersil ODS, das Ergebnis lautet: Eine hervorragende Selektivität für die zwei starke Basen bei gleichzeitig sehr langsamen Kinetik (starkes Tailing). Dass weitere polare Phasen wie beispielsweise eine C7-fluorierte Phase eine ebenso gute Selektivität aufweist (siehe mittleres Chromatogramm) versteht sich von selbst.
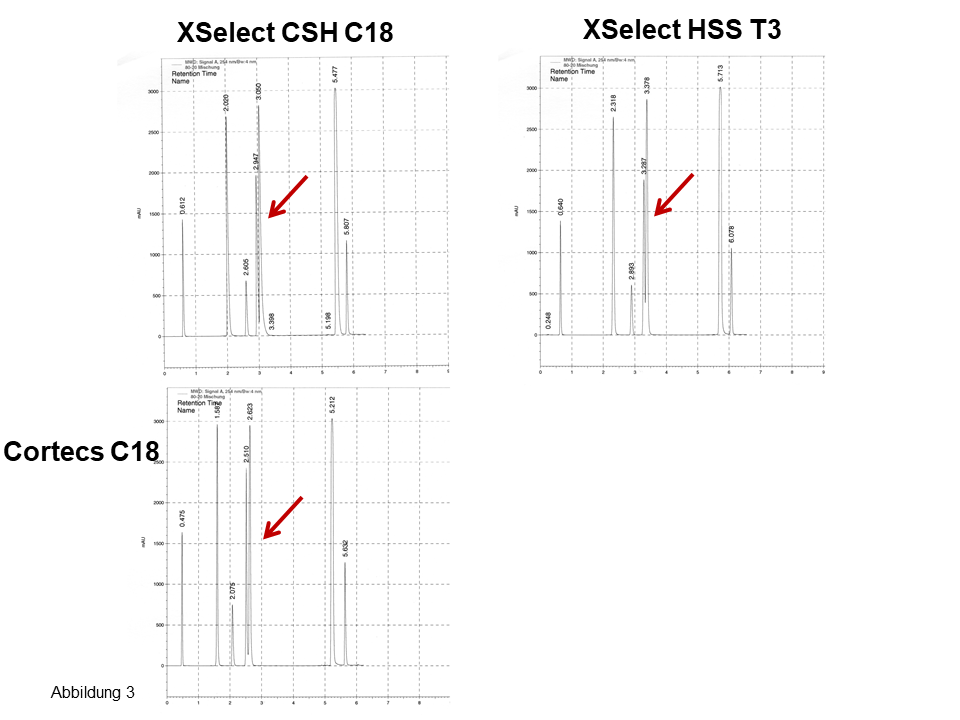
Beispiel 3: Injektion einer Mischung diverser Komponenten inkl. drei Isomeren (o-, m-, p-Toluidin)
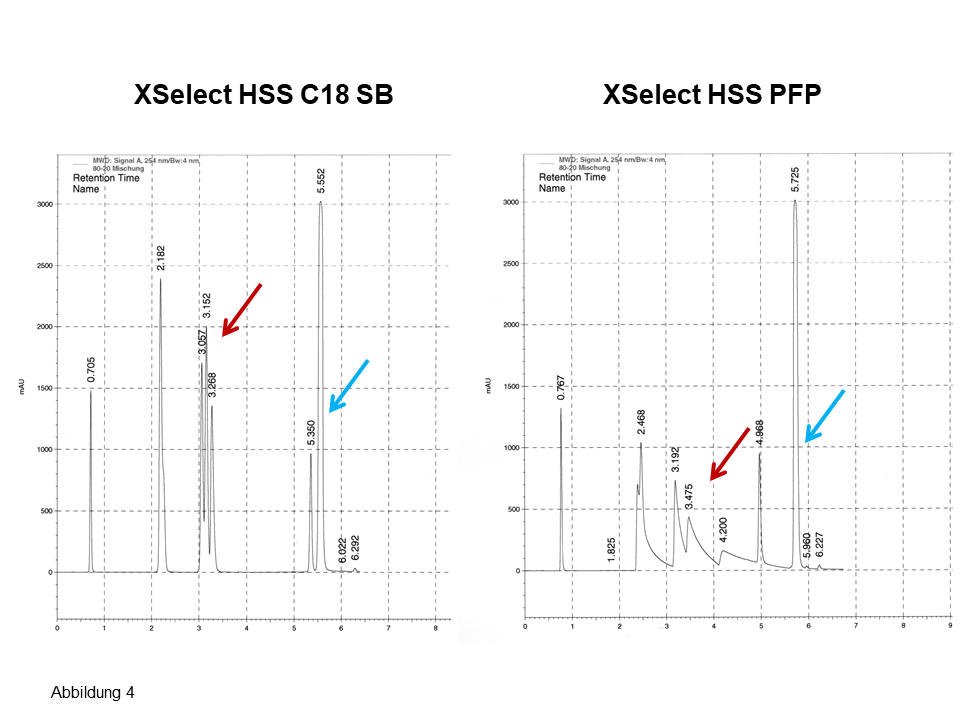
An mehreren Säulen von Waters erhält man nahezu das gleiche Bild, die Chromatogramme sehen recht ähnlich aus, für die drei Isomere ergeben sich zwei Peaks, siehe Pfeile in Abbildung 3. Erst beim Einsetzen einer nicht-endcappeden Phase (Abbildung 4) sind für die drei Isomere drei Peaks zu sehen. Ferner: Betrachte bei den letzten zwei Peaks die Elutionsumkehr. Auch hier: Eine fluorierte Phase (Abbildung 4, rechts) zeigt eine noch bessere polare Selektivität bei einer noch langsameren Kinetik, siehe dazu das auffallend starke Tailing.
Abb. 3 Trennung von polaren und apolaren Aromaten inkl. Stellungsisomeren, Erläuterung, siehe Text
Abb. 4 Trennung von polaren und apolaren Aromaten inkl. Stellungsisomeren, Erläuterung, siehe Text
Das Fazit
Für eine Vielzahl üblicher Trennprobleme sind moderne, endcappede Materialien zweifelsohne die richtige Wahl. Es gibt jedoch Fälle, in denen gerade Restsilanolgruppen eine Erhöhung der Selektivität bedingen. Das ist generell der Fall, wenn für die Selektivität zusätzliche Ionenaustausch-Wechselwirkungen notwendig sind wie beispielsweise bei Stellungsisomeren, Phospholipiden und starken Säuren/Basen. Eine evtl. suboptimale Peakform muss oft zu Gunsten einer guten polaren Selektivität in Kauf genommen werden. Es gilt folgende vereinfachte Regel: Je ähnlicher die Moleküle sind, umso notwendiger sind zusätzliche polare/ionische Wechselwirkungen für deren Trennung, umso langsamer die Kinetik bei der Desorption solcher Moleküle von der stationären Phase. Zur Auswahl und zum Vergleich von RP-Säulen siehe „Colona Vergleich und Auswahl von HPLC-RP-Säulen“ , ferner auch das Dokument „Einfache Tests zur Charakterisierung von HPLC-RP-Säulen“.
Der Fall In diesem HPLC-Tipp haben wir uns über folgenden Tatbestand unterhalten: Wenn ich in der HPLC einen physikalischen Parameter verändere, ist das Ergebnis selten eindeutig „gut“ oder „schlecht“. Je nach Betrachtungsweise bzw. Anforderungen überwiegen Vor- oder eben Nachteile. Dies gilt analog auch für chemische Parameter. Hier wollen wir stellvertretend sechs solcher betrachten, siehe Tabelle 1. Die Lösung Was Nachteile Vorteile Erhöhung der Temperatur In der Regel Abnahme der Selektivität und der Lebensdauer der Säule, ferner erhöhte Gefahr für Zersetzung von thermolabiler Analyten Abnahme der Retentionszeit und des Druckes, Verbesserung der Peakform und dadurch Zunahme der Peakkapazität (Anzahl der…