Säulendimensionen und Totvolumen: Wie beeinflussen sie die Trennung im Gradienten-Modus im Vergleich zu isokratischen Trennungen?
Zusammenfassung:
Isokratische Methoden:
Bei großen Säulenvolumina spielt das Totvolumen eine untergeordnete Rolle, bei kleinen Säulenvolumina (kurze/dünne Säulen) sehr wohl, vereinfacht gesagt: Eine lange/dicke Säule „verzeiht“ eine schlechte Apparatur, eine kurz/dünne tut es nicht.
Gradienten-Methoden:
Das Totvolumen spielt hier kaum eine erwähnenswerte Rolle.
Erläuterungen und Beispiele:
In Abbildung 1 wird eine isokratische Trennung an zwei Hypersil GOLD-Säulen gezeigt; links eine 125 x 4 mm-Säule, rechts eine 50 x 2,1 mm-Säule.
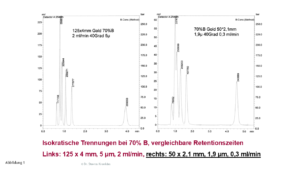
Abbildung 1
Isokratische Trennung an einer großen (links) und an einer kleinen (rechts) Säule, Details, siehe Text
Die Auflösung der fünf Peaks an der kleineren Säule (rechts) ist schlechter, die Peaks sind recht breit. Wir brauchen keine Formeln, um das zu erklären, ein einfacher Zahlenvergleich reicht aus:
– Die große Säule hat ein Säulenvolumen von ca. 1.250 µl, die kleine von ca.139 µl
– Das Totvolumen der Apparatur (Volumen vom Autosampler bis einschließlich
Detektor ohne Säule) betrug hier ca. 80 µl
Das Totvolumen entspricht somit nur ca. 6,4 % dem Säulenvolumen der großen Säule.
Bei der kleinen Säule sind es ca. 58,0 % – und das ist nicht grade wenig …
Halten wir fest: Je kleiner das Säulenvolumen ist, umso kritischer wird bei isokratischen Trennungen das Totvolumen der Apparatur.
In Abbildung 2 ist ein Gradientenlauf mit der kleinen Säule an der gleichen Apparatur zu sehen: Wir erhalten eine fantastische Peakform – trotz des weiterhin vorhandenen recht großen Totvolumens.
Im Gradientenmodus spielt das Totvolumen der Apparatur kaum eine Rolle:
Die während eines Gradientenlaufs immer stärker werdende Elutionskraft (immer mehr % B) sorgt dafür, dass die Moleküle an der hinteren Flanke des Peaks stärker „nach vorne“ geschoben werden als die Moleküle an der vorderen Flanke. Dies wirkt gegen die in Flussrichtung stets zunehmende Bandenverbreiterung aus, Ergebnis: Die zwei Effekte heben sich gegenseitig in etwa auf, die Peaks bleiben beim Gradienten schmal. Das ist auch der Grund, warum beim Gradienten die Peakbreite aller Peaks im Idealfall gleich ist.

Abbildung 2
Gradiententrennung mit einer kleinen Säule an einer „schlechten“ Apparatur, Details, siehe Text
Ein zweites, schönes Beispiel von Monika Dittmann, Agilent, dient, um das bereits Vorgestellte noch einmal zu veranschaulichen: In Abbildung 3 oben (A) wird eine isokratische Trennung mit einer 1-mm-Säule gezeigt, hier liegt also ein recht kleines Säulenvolumen vor. Durch eine bewusst falsch angeschlossene Kapillare entstand ein merkliches Totvolumen, die Peaks zeigen ein starkes Tailing. Ohne an der Apparatur etwas zu ändern, wurde mit der gleichen Säule ein Gradient gefahren, Abbildung 3, Mitte (B). Ergebnis: Hervorragende Peakform. Durch eine korrekte Kapillarverbindung und dadurch Eliminierung des Totvolumens ergab sich nun anschließend auch im isokratischen Modus eine sehr gute Peakform, Abbildung 3, unten (C).
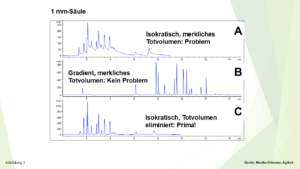
Abildung 3
Isokratische und Gradienten-Trennungen mit einer 1 mm-Säule mit und ohne merklichem Totvolumen der Apparatur, Details, siehe Text
Eine Bemerkung, bevor wir zum Fazit kommen:
Ich spreche hier vom „Totvolumen“, weil dieser Begriff geläufig ist und als erste Näherung den Sachverhalt genügend gut beschreibt. Genauer müsste man/frau vom Dispersionsvolumen sprechen: Eine lange Kapillare mit einem kleinen Durchmesser mag das gleiche (Tot)Volumen aufweisen wie eine kürzere mit einem größeren Durchmesser. Das Dispersionsvolumen aber, das vom Durchmesser in der 4. Potenz abhängt, ist im ersten Fall viel kleiner. Und da die Peakverbreiterung vom Dispersionsvolumen beeinflusst wird, wäre die Peakform im ersten Fall wesentlich besser.
Schlussfolgerung:
Nehmen Sie lieber eine lange, dünne Kapillare statt einer kurzen, dicken.
Fazit:
Wenn Sie an einer Apparatur ausschließlich im Gradienten-Modus arbeiten, entsteht für „normale“ Fragestellungen kaum Handlungsbedarf bzgl. Hardwareoptimierung – alle Gradienten-Anlagen sind gut genug. Arbeiten Sie an besagter Apparatur auch isokratisch, müssten Sie bei Verwendung von kurzen/dünnen Säulen und kleinen Teilchen evtl. tätig werden, z. B. an dünnere Kapillaren, kleineres Zellvolumen, totvolumenfreie Verbindungen und/oder besseres Design der Detektor-Zelle denken. Sonst verlieren Sie unter Umständen an Trennleistung.

