Heute: Wie beeinflusst die Mischkammer meine Trennung? Der Gradient in der HPLC hat wahrlich eine Menge interessanter Aspekte … Ich greife für die nächsten sechs HPLC-Tipps einen einzigen Aspekt heraus: Was genau können physikalische Parameter beim Gradienten bewirken? Und: Was ist dabei anders im Vergleich zu isokratischen Trennungen? Unter „physikalische“ Parameter sind folgende gemeint, die wir uns heute und in den nächsten HPLC-Tipps näher anschauen werden: Mischkammer Totvolumen bzw. Verweil- oder Verzögerungsvolumen Säulendimensionierung und Kapillarlänge Fluss Teilchengröße Heute geht es um die Mischkammer: Volumen sowie Geometrie/Design Zusammenfassung: Das Volumen aber auch die Geometrie/Design der Mischkammer (Mischer, Mischventil) können einiges beeinflussen: Rauschen und „Buckel“ in der Basislinie, Retentionszeit, Auflösung, Anzahl der Peaks, Auftauchen von Geisterpeaks. Mischkammer-Volumen und Rauschen Ein größeres Mischkammer-Volumen führt in der Regel zu einer besseren Durchmischung der Lösungsmittel und folglich zu einem geringeren Rauschen. Das macht sich vor allem in folgenden Fällen bemerkbar: Bei niedrigen Wellenlängen, bei quaternären Pumpen (Niederdruck-Gradient) und bei komplexen Eluenten, die beispielsweise Modifier enthalten (z. B. IP-RP für Oligos). In Abbildung 1 wird das Rauschen bei einer 35 µl- vs. 135 µl- und in Abbildung 2 das Rauschen bei einer 50 µl- vs. 340 µl-Mischkammer gezeigt Abbildung 1 Basislinie bei einer 35 µl- und…
Der Fall Im HPLC-Tipp vom letzten Dezember war Peaky am Zweifeln, ob er es wirklich ist: „Bin ich überhaupt der Peaky? Und wenn ja, wieso fühlt es sich so an, als wäre ich mehrere …“ Es geht also um die Frage, ob ein Peak homogen ist oder womöglich doch eine Koelution vorliegt. Im vorliegenden HPLC-Tipp werden wir uns einfache Tests anschauen, die recht schnell durchzuführen sind: Keine Änderung der chromatographischen Parameter, keine Änderung der Apparatur. In den nächsten HPLC-Tipps werden wir uns sukzessiv mit aufwendigeren Methoden beschäftigen. Die Lösung Eine Veränderung von Einstellparameter („Settings“) kann helfen, innerhalb von Sekunden/Minuten die Peakhomogenität zu überprüfen. Es lohnt sich (auch) an solche „banale“, schnelle Möglichkeiten zu denken: Injektion bei einer anderen (niedrigeren) Wellenlänge, siehe Abbildung 1: Bei 271 nm erscheint der dritte Peak als ein Peak (unteres Chromatogramm), bei 225 nm sind zwei Peaks zu erkennen (oberes Chromatogramm) 2. Eine große Zeitkonstante („Filter Response“, „Filter Time Constant“) führt zwar zu einer ruhigeren Basislinie, die Peakbreite nimmt allerdings zu, man verliert an Auflösung. Folgendes Zahlenbeispiel aus einer „Technical Note“ von Waters, die freundlicherweise von Sascha Schifrin zur Verfügung gestellt wurde: Kein digitales Filtern: Auflösung (Resolution) 3,16, Peakkapazität 16 Zeitkonstante, 0,5 Min: Auflösung 1,82,…
Der Fall Nehmen wir an, Sie haben recht viele, recht ähnliche Komponenten zu trennen. In einem solchen Fall ist eine ausreichende Selektivität – also unterschiedlich starke Wechselwirkungen der einzelnen Komponenten mit der stationären Phase – realistischerweise kaum erreichbar. Der einzige Ausweg lautet: Eine möglichst gute Peakkapazität, also maximal mögliche Anzahl Peaks pro Zeiteinheit. Wie ist dies zu erzielen? Die Lösung Es gibt mehrere Formeln für die Peakkapazität, die zwei einfachsten sind folgende: nC = tRl – tRf / w und nC = tG / w mit: nC: Peakkapazität tRl: Retentionszeit des letzten Peaks tRf: Retentionszeit des ersten Peaks tG: Gradientendauer w: Peakbreite Was heißt das nun? Vereinfacht folgendes: Ich brauche eine große Differenz zwischen der Retentionszeit des letzten und des ersten Peaks und die Peakbreite soll möglichst klein sein. Diese allgemeine Forderung ist „zeitlos“, gilt für alle Gradientenarten und ist auch unabhängig davon, ob es sich um kleine oder große (Bio)Moleküle handelt. D. h. sie ist anwendbar sowohl imfalle von RP-Trennungen als auch beispielsweise bei Ionenaustauschertrennungen von Oligonucleotiden mittels Salz- bzw. pH-Wert-Gradienten. Was braucht man also? * Einen langen Gradienten und einen hohen Fluss (großes Gradientenvolumen) * Eine lange, möglichst dünne Säule (große Retentionszeitdifferenz letzter/ertster Peak, maximal erreichbare Auflösung aufgrund…
1. „Schwach“ ist oft effektiver – im positiven oder im negativen Sinn 2. Auflösung bei einer RP-Trennung nicht vorhanden – effektive Maßnahmen „Schwach“ ist oft effektiver – im positiven oder im negativen Sinn An schwachen Ionenaustauscher (WCX) werden sehr ähnliche ionische Komponenten häufig besser getrennt als an starken (SCX) Mit einer schwachen Probelösung(im RP-Modus: Jene einfach mit Wasser verdünnen oder mit Neutralsalz versetzen) erreicht man häufig eine Verbesserung der Peakform und somit der Auflösung, vor allem bei früh eluierenden Peaks Analog die mobile Phase: Ein schwächerer Eluent (im RP-Modus: Wasser-/Pufferreich) führt in der Regel zu einer besseren Trennung Eine schwächere Isopropanol/Wasser-Lösung (z. B….
Permanent auftretender Geisterpeak Faustregeln zu Log P und Log D Abnahme der Auflösung – eine mögliche Ursache Seit kurzem erscheint ein Geisterpeak bzw. stets ein Blindwert im Blank… Seit einiger Zeit taugt bei einer Methode – oder bei mehreren – die noch vor kurzem keine Probleme machte(n), ein Geisterpeak auf. Oder aber, Sie haben in Ihrem Blank auf einmal stets einen Blindwert. Und dieses Problem haben Sie unabhängig von der Anlage, dem/der Anwender(in) und sogar der Methode. D.h. das übliche Fehler-Ausschluss-Programm haben Sie gründlich aber leider erfolglos absolviert. Versuchen Sie in einem solchen Fall im Team sich zu erinnern,…
Der Fall Sie arbeiten in einem regulierten Bereich und sind an strenge Prüfvorschriften gebunden. In einer solchen lautet die Forderung: „Auflösung R größer 1,5“, aber gerade diesen Wert erreichen Sie aktuell nicht. Welche regel-konforme Handgriffe kämen in Frage? Die Lösung Was möglich ist, hängt letzten Endes davon ab, wie genau die Angaben in der betreffenden PV sind. Ist in der Tat restlos alles – von der Eluentenzusammensetzung bis zu den Einstellungen („Settings“) – vorgegeben, so können Sie de facto es nur mit einer neuen Säule versuchen. Ist die PV etwas „weicher“, d.h. es sind nur die wichtigsten Parameter wie Säule,…
In den letzten Jahren wurde eine Reihe moderner C18- sowie polarer RP-Phasen eingeführt. Da wären beispielhaft zu nennen: Chemisch geschützte („embedded“ phases), hydrophil endcappede sowie Mixed Mode Phasen und was die Matrix betrifft: Hybrid-, Core Shell- oder monolithische Phasen. Diese Materialien weisen vielfach Vorteile auf. Heißt es nun, dass bei der Entwicklung einer neuen Methode der Einsatz einer solchen modernen Phase die richtige Wahl wäre? Sollte man also nicht-endcappede Phasen als eine alte Technologie „ad acta“ legen?
Die Lösung
Nein. Für die Trennung von Substanzen mit ähnlicher Hydrophobie, aber mit Unterschieden in der Anordnung von Substituenten am Molekül oder von Doppelbindungen in einer Seitenkette (α, β-Isomerie, Stellungsisomerie) oder stark polare Komponenten sind Restsilanolgruppen für die Selektivität sehr wichtig. Dies wird an drei Beispielen demonstriert:
Beispiel 1: Trennung von Steroiden
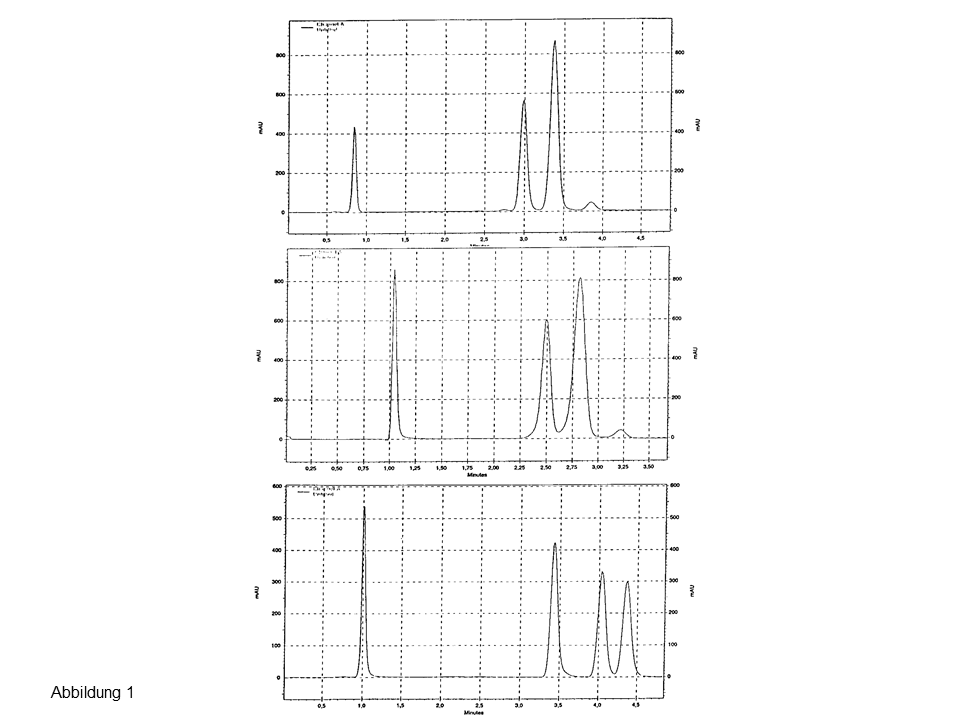
Abb. 1: Trennung von drei Steroiden an zwei endcappeden (oben, Mitte) und an einer nicht endcappeden C18-Phase (unten), Erläuterungen siehe Text.
Das obere und mittlere Chromatogramm zeigen die Injektion von drei Steroiden (α, β-Isomere) an zwei modernen, hydrophoben Phasen. Steroid Nr. 2 und 3 koeluieren. Die Trennung gelingt an Resolve C18, einem älteren, nicht endcappeden Material, siehe unteres Chromatogramm in Abbildung 1.
Beispiel 2: Trennung von starken Basen
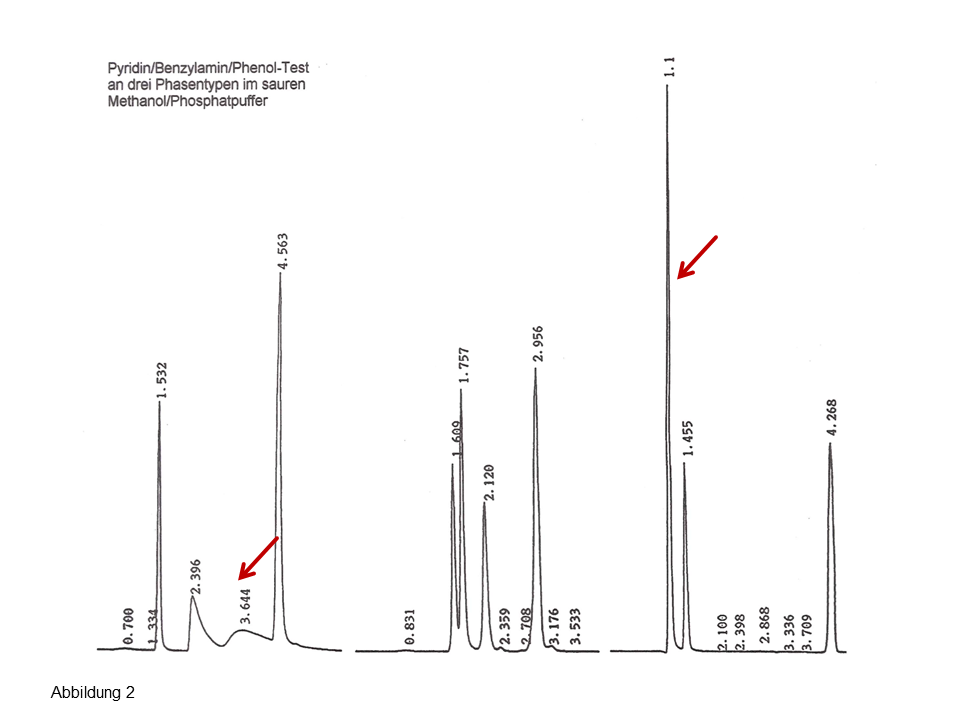
Abb. 2: Injektion einer Mischung von drei polaren Komponenten auf eine silanophile (links) und eine hydrophobe, endcappede C18-Phase (rechts), Erläuterungen, siehe Text.
Auf der rechten Seite der Abbildung 2 wird die Injektion von Uracil (inerte Komponente), Pyridin, Benzylamin und Phenol an einer modernen endcappeden C18-Säule gezeigt. Die zwei Basen koeluieren (erster Peak), was vollkommen nachvollziehbar ist: Man kann nicht erwarten, dass eine hydrophobe, gründlich endcappede Phase eine gute polare Selektivität aufweist. Und das kann zu falschen Schlussfolgerungen führen: Eine gute Peaksymmetrie suggeriert im Routinealltag eine gute Selektivität… Das linke Chromatogramm zeigt die Injektion auf eine „alte“, stark silanophile Phase, Hypersil ODS, das Ergebnis lautet: Eine hervorragende Selektivität für die zwei starke Basen bei gleichzeitig sehr langsamen Kinetik (starkes Tailing). Dass weitere polare Phasen wie beispielsweise eine C7-fluorierte Phase eine ebenso gute Selektivität aufweist (siehe mittleres Chromatogramm) versteht sich von selbst.
Beispiel 3: Injektion einer Mischung diverser Komponenten inkl. drei Isomeren (o-, m-, p-Toluidin)
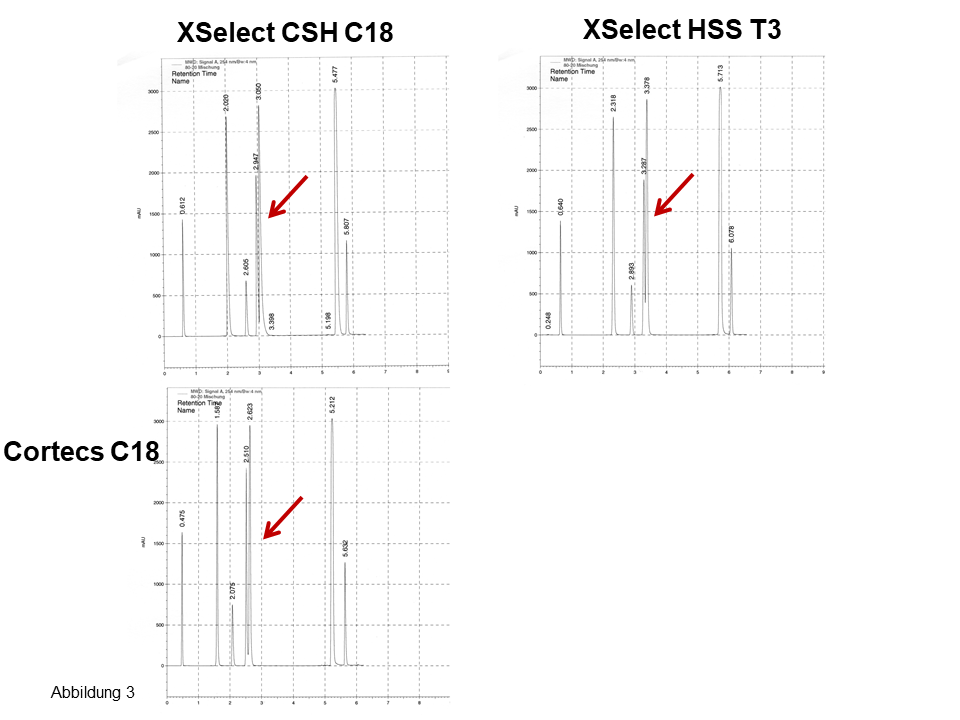
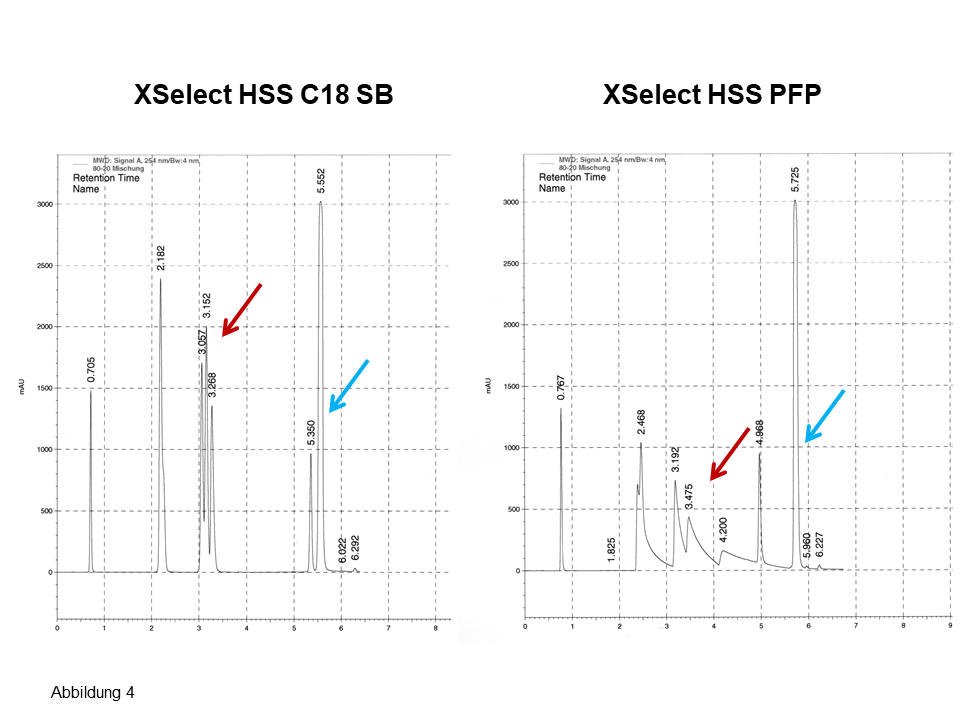
An mehreren Säulen von Waters erhält man nahezu das gleiche Bild, die Chromatogramme sehen recht ähnlich aus, für die drei Isomere ergeben sich zwei Peaks, siehe Pfeile in Abbildung 3. Erst beim Einsetzen einer nicht-endcappeden Phase (Abbildung 4) sind für die drei Isomere drei Peaks zu sehen. Ferner: Betrachte bei den letzten zwei Peaks die Elutionsumkehr. Auch hier: Eine fluorierte Phase (Abbildung 4, rechts) zeigt eine noch bessere polare Selektivität bei einer noch langsameren Kinetik, siehe dazu das auffallend starke Tailing.
Abb. 3 Trennung von polaren und apolaren Aromaten inkl. Stellungsisomeren, Erläuterung, siehe Text
Abb. 4 Trennung von polaren und apolaren Aromaten inkl. Stellungsisomeren, Erläuterung, siehe Text
Das Fazit
Für eine Vielzahl üblicher Trennprobleme sind moderne, endcappede Materialien zweifelsohne die richtige Wahl. Es gibt jedoch Fälle, in denen gerade Restsilanolgruppen eine Erhöhung der Selektivität bedingen. Das ist generell der Fall, wenn für die Selektivität zusätzliche Ionenaustausch-Wechselwirkungen notwendig sind wie beispielsweise bei Stellungsisomeren, Phospholipiden und starken Säuren/Basen. Eine evtl. suboptimale Peakform muss oft zu Gunsten einer guten polaren Selektivität in Kauf genommen werden. Es gilt folgende vereinfachte Regel: Je ähnlicher die Moleküle sind, umso notwendiger sind zusätzliche polare/ionische Wechselwirkungen für deren Trennung, umso langsamer die Kinetik bei der Desorption solcher Moleküle von der stationären Phase. Zur Auswahl und zum Vergleich von RP-Säulen siehe „Colona Vergleich und Auswahl von HPLC-RP-Säulen“ , ferner auch das Dokument „Einfache Tests zur Charakterisierung von HPLC-RP-Säulen“.
Der Fall Additiva (Modifier) oder einfache Zusätze wie Salze in der mobilen Phase können die Trennung positiv beeinflussen, ihr Einsatz bewährt sich seit Jahrzehnten. So kann beispielsweise die Peaksymmetrie und dadurch indirekt die Empfindlichkeit durch Verwendung von etwa Di- oder Triethylamin, DEA/TEA, Di-Tri- oder Fluoressigsäure, DFA/TFA/FA, Ionenpaarreagenzien wie Heptan- oder Oktansulfonsäure erhöht werden. Das Problem dabei: Solche Additiva werden teilweise an der Oberfläche der stationären Phase (irreversibel) adsorbiert, was in der Routine vielleicht ein geringe(re)s Problem darstellt. In der Methodenentwicklung jedoch ist dies störend, weil ja nach jedem Experiment die Säule mühsam durch Spülen wieder in den ursprünglichen Zustand gebracht…
Der Fall Mithilfe von SSTs wird überprüft, ob diese Methode an diesem Gerät „hier und jetzt“ funktioniert. Ob also die Anforderungen an diese Methode erfüllt werden oder eben nicht. Als Forderung kann beispielsweise ein bestimmter Wert für die Auflösung eines kritischen Paars oder die Bestimmungsgrenze für eine Nebenkomponente gelten. Sind weitere Tests notwendig zumal ja die Zeit immer knapper wird? Die Lösung Sicherlich gibt es hier unterschiedliche Vorstellungen. Nachfolgend meine persönliche Meinung und davon ableitend ein Vorschlag: Ein gut designter SST beinhaltet die Information, die ich brauche. Passt er, so ist nichts mehr zu tun. Passt er nicht, so wären…
Der Fall Es gibt in der HPLC mehrere Möglichkeiten, die Peaks nach vorne zu schieben. Dabei rücken sie zusammen, die Auflösung nimmt natürlich ab. So lautet eine (vor)schnelle Schlussfolgerung. Das stimmt allerdings nicht immer. Wann nicht und warum? Die Lösung Betrachten wir hier ein klassisches RP-System. Ich kann die Wechselwirkungen zwischen Analyten und stationärer Phase wie folgt verringern, die Peaks eluieren dabei früher: Den organischen Anteil der mobilen Phase bzw. die Temperatur erhöhen, den pH-Wert verändern, schließlich kann ich eine polarere stationäre Phase verwenden. Wenn meine Substanzen nun sich chemisch ähneln und dementsprechend auch chromatographisch ähnlich verhalten – man spricht…