Heute: Wie beeinflusst die Mischkammer meine Trennung?
Der Gradient in der HPLC hat wahrlich eine Menge interessanter Aspekte …
Ich greife für die nächsten sechs HPLC-Tipps einen einzigen Aspekt heraus: Was genau können physikalische Parameter beim Gradienten bewirken? Und: Was ist dabei anders im Vergleich zu isokratischen Trennungen? Unter „physikalische“ Parameter sind folgende gemeint, die wir uns heute und in den nächsten HPLC-Tipps näher anschauen werden:
- Mischkammer
- Totvolumen bzw. Verweil- oder Verzögerungsvolumen
- Säulendimensionierung und Kapillarlänge
- Fluss
- Teilchengröße
Heute geht es um die Mischkammer: Volumen sowie Geometrie/Design
Zusammenfassung:
Das Volumen aber auch die Geometrie/Design der Mischkammer (Mischer, Mischventil) können einiges beeinflussen: Rauschen und „Buckel“ in der Basislinie, Retentionszeit, Auflösung, Anzahl der Peaks, Auftauchen von Geisterpeaks.
Mischkammer-Volumen und Rauschen
Ein größeres Mischkammer-Volumen führt in der Regel zu einer besseren Durchmischung der Lösungsmittel und folglich zu einem geringeren Rauschen. Das macht sich vor allem in folgenden Fällen bemerkbar: Bei niedrigen Wellenlängen, bei quaternären Pumpen (Niederdruck-Gradient) und bei komplexen Eluenten, die beispielsweise Modifier enthalten (z. B. IP-RP für Oligos). In Abbildung 1 wird das Rauschen bei einer 35 µl- vs. 135 µl- und in Abbildung 2 das Rauschen bei einer 50 µl- vs. 340 µl-Mischkammer gezeigt
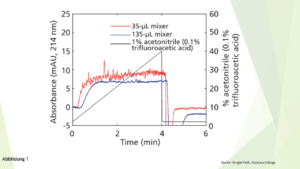
Abbildung 1
Basislinie bei einer 35 µl- und einer 135 µl-Mischkammer

Abbildung 2
Basislinie bei einer 50 µl- und einer 340 µl-Mischkammer
Mischkammer-Volumen und Auflösung
Durch ein verändertes Volumen der Mischkammer ändert sich das Verweilvolumen (Volumen von der Mischkammer bis zum Säulenkopf). Und dies hat einen Einfluss auf viele Sachen, u. a. auf die Auflösung. Und weil der Gradient eben recht „tricky“ ist, leider nicht immer einheitlich. In Abbildung 3 wird die Trennung mit einer 1,7 ml-Mischkammer gezeigt: Die Trennung im vorderen Bereich des Chromatogramms ist einigermaßen OK, der Hauptpeak bei ca. 4,6 min sieht ordentlich aus. Bei Verwendung einer 2,6 ml-Mischkammer (Abbildung 4) ist die Trennung zwar vorne schlechter, dafür erscheinen beim Hauptpeak Verunreinigungen, es ergibt sich also just dort eine Verbesserung der Auflösung. Die Peakform von Peak 1 und 2 bleibt in beiden Fällen unverändert.

Abbildung 3
Trennung mit einer 1,7 ml-Mischkammer, Details, siehe Text

Abbildung 4
Trennung mit einer 2,6 ml-Mischkammer, Details, siehe Text
Mischkammer-Volumen und Design
Wichtig ist nicht lediglich das Volumen der Mischkammer, sondern auch das Design. Abbildung 5 zeigt eine Trennung mit der Original-400 µl-Mischkammer von Agilent, Abbildung 6 mit einer 10 µl „Lee-Mischkammer“. Obwohl im zweiten Fall das Volumen um Faktor 40 kleiner ist, wird eine nur minimal schlechtere Auflösung im vorderen Teil des Chromatogramms beobachtet. Das heißt: Behalte im Blick nicht nur das Volumen der Mischkammer, ein ausgeklügeltes Design ist mindestens so wichtig für eine homogene Durchmischung der Lösungsmittel: Eine kleine „Säule“ mit Glaskugeln, ein Mischer aus Siebteilchen, ein „Würfel“ mit irregulären Metallstäbchen usw. Übrigens: Das Design entscheidet auch, inwieweit Polymere am Eingang zur Mischkammer entstehen, falls rein ACN gefördert wird; ob also „Buckel“ in der Basislinie erscheinen und ob sich evtl. Druckschwankungen ergeben.
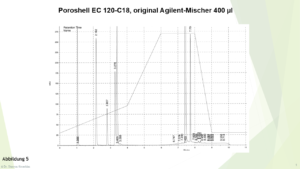
Abbildung 5
Auflösung bei Verwendung einer Agilent-400 µl-Mischkammer, Details, siehe Text
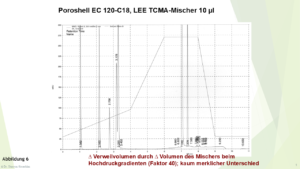
Abbildung 6
Auflösung bei Verwendung einer 10 µl-„Lee-Mischkammer“, Details, siehe Text
Mischkammer-Volumen und Retentioszeit, Anzahl der Peaks, Geisterpeaks
In Abbildung 7 wird ein schönes Beispiel von Jürgen Maier-Rosenkranz gezeigt: Zunächst der programmierte Gradient, ferner der tatsächlich resultierende, einmal mit einer 1 ml- und einmal mit einer 2 ml-Mischkammer.
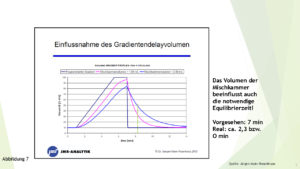
Abbildung 7
Programmierter und tatsächlicher Gradient mit einer 1 ml- bzw. 2 ml-Mischkammer, Details, siehe Text
Nachfolgend zusammengefasst, erstens was passiert und zweitens was dies in der Praxis bedeutet:
– Die vorgesehene (programmierte) Spülzeit von 2 min findet aufgrund der
Verzögerung des Gradienten durch das Volumen der Mischkammer gar nicht statt.
Die Säule wird am Ende des Gradienten demnach gar nicht gespült
– Geringere Elutionskraft am Ende des Gradienten; mit der 1 ml-Mischkammer,
ca. 97 % B und mit der 2 ml-Mischkammer ca. 85 % B; es könnte also passieren,
dass dadurch nicht alle Verunreinigungen von der stationären Phase herunter
gespült werden
– Kürzere Equlibrierzeit als die vorgesehene von 7 min: Mit der 1ml-
Mischkammer nur ca. 2,5 min bzw. überhaupt keine Equlibrierzeit im Falle der
2 ml-Mischkammer. Das bedeutet, die nächste Injektion erfolgt, bevor die
Säule im Gleichgewicht gewesen ist.
Merke somit: Bei sonst gleichen Anlagen aber unterschiedlichen Mischkammern müsste man/frau u. U. mit folgendem rechnen – neben einer Veränderung der Auflösung und Unterschieden in der Basislinie/im Rauschen: Verschiebung der Retentionszeit, mehr Peaks, Erscheinen von Peaks beim nächsten Lauf.

