Liebe Leserinnen, liebe Leser,
weiter unten finden Sie den jeweils ersten Teil der halben Sätze aus dem Dezember-Tipp, so dass nun die Befunde komplett sind:
Ich weiß, dass …
… sich der Fluss geändert hat (Leck, Luft in der Pumpe)… weil die Peakfläche, aber kaum die Peakhöhe sich geändert hat
… das Probelösungsmittel organischer ist als der Eluent/der Anfangsgradient… weil die frühen Peaks mit einem starken Fronting eluieren
… der pH-Wert des Eluenten sich geändert hat oder – seltener – funktionelle Gruppen auf der stationären Phase hydrolysiert worden und dadurch nun mehr Silanolgruppen vorhanden sind … weil nur einige Peaks tailen und auch später eluieren – restliche Peaks sind OK
… dass ich eine recht hydrophobe, endcappte, „klassische“ C18 stationäre Phase habe… weil eine derartige RP-Phase sehr ähnliche Komponenten (z. B. Isomere) nicht trennen kann
… dass ich in der Probe entweder sehr große Moleküle oder sehr kleine, ionisch vorliegende Substanzen habe… weil solche Komponenten vor der Totzeit eluieren (Peaks vom letzten Lauf, Kontaminationen im Eluenten, Luft usw. können ausgeschlossen werden)
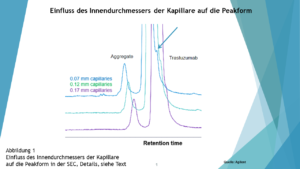
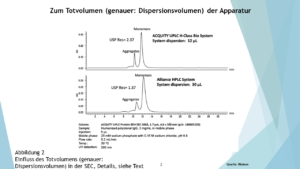
… dass das Totvolumen der Apparatur zu groß im Vergleich zu dem Säulenvolumen ist…weil dies die einzige Ursache sein kann, dass bei neutralen Komponenten die Peaksymmetrie mit zunehmender Retention besser wird
— wenn es um das Erfassen aller Komponenten in der Probe geht, zusätzlich ein Universaldetektor wie z. B. CAD (Charge Aerosol Detector) benötigt wird… weil nicht alle Substanzen UV-aktiv sind bzw. ionisiert werden können
… ich Methanol als organisches Lösungsmittel und niedrige Temperatur brauche …weil bei diesen Bedingungen polare Komponenten in der Regel besser zu trennen sind
…meine Moleküle mit ausgewaschenen Metallionen aus allerlei Metalloberflächen Komplexe bilden bzw. dort teilweise adsorbiert werden… weil bei Verwendung von PEEK-Kapillaren, einer PEEK-lined Säule sowie einer Injektionsnadel aus PEEK oder Keramik die Peaksymmetrie wesentlich besser wurde
…ich meine wässrige Probelösung mit 5-10% ACN oder Iso-OH versetzen sollte …weil durch diesen Zusatz in der wässrigen Probelösung die Adsorption der Analyte an allerlei Metalloberflächen in der HPLC-Apparatur (Physisorption, Adhäsion) verhindert bzw. minimiert wird